

What Is ICH GCP Certification 2024
What Is ICH GCP Certification?
ICH, which stands for the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, is a pivotal global organization shaping the landscape of clinical trials in 2024. Responsible for setting stringent standards, ICH ensures that clinical trials involving human subjects adhere to rigorous regulatory frameworks.
In essence, ICH oversees the implementation of Good Clinical Practice (GCP) guidelines, ensuring that the planning, execution, and documentation of clinical trials are conducted ethically and with scientific integrity at every phase of the research process.
For those seeking to enhance their understanding of ICH GCP guidelines, there's an opportunity to access free online training, providing invaluable insights into the intricacies of conducting ethical and scientifically sound clinical trials in 2024.
You can demo ich gcp training free online here.
What is the purpose of GCP Certification?
In 2024, GCP Certification, short for Good Clinical Practice Certification, serves as a crucial acknowledgment of an individual's proficiency and expertise in implementing regulatory guidelines across all facets of their work.
Why is it significant?
In the realm of clinical research, possessing GCP certification is indispensable for success. It's not just a matter of preference; it's a necessity. Any organization involved in clinical research endeavors must ensure that their personnel comprehensively grasp the ICH guidelines and are certified to conduct scientific studies accordingly. In fact, many companies provide certification programs for their employees even before they commence their duties. This underscores the paramount importance of GCP certification in the clinical research industry landscape of 2024.ICH GCP Attestation Form
If you're looking to explore a career in the innovative and quickly growing field of clinical research, you'll need to ensure you get the proper Good Clinical Practice certification.
GCP Certification
Are you seeking a GCP refresher online course or comprehensive initial advanced GCP training in 2024? Look no further. Our cutting-edge GCP certification courses are designed to equip you with all the necessary tools to obtain your certification efficiently.
In today's fast-paced world, attending in-person classes for GCP certification training may not always be feasible. That's why our innovative and user-friendly online courses offer a convenient alternative. We understand the importance of receiving top-notch training from experts well-versed in the ICH GCP guidelines to propel your career forward.
At CCRPS, excellence is our standard. Each ICH GCP module we offer is crafted with meticulous attention to detail, ensuring that you receive the highest level of instruction possible. It's time to invest in yourself and receive the refresher training you deserve from professionals who understand how to facilitate your advancement.
Whether you're an ethics committee member, clinical research staff member, or a student embarking on a career in clinical research, our training programs are tailored to meet your needs and set you up for success in the dynamic field of clinical research in 2024.
Good Clinical Practice Training Certificate
How can CCRPS propel your career in 2024?
At CCRPS, we're dedicated to equipping individuals in or aspiring to join the clinical trial industry with the necessary knowledge and training conveniently accessible at their fingertips. We understand that committing hours to classroom study isn't always feasible amidst your professional responsibilities. That's why we've developed our comprehensive GCP refresher course, designed to be completed entirely online.
Enrolling in our course offers you the opportunity to delve into advanced-level content to prepare for your Good Clinical Practice Certification testing.
What benefits await you upon enrollment in our practice training?
While GCP certification is mandatory for all clinical research professionals, its significance is particularly pronounced for certain groups:
Investigators representing drug companies, research centers, hospitals, and more.
Members of ethics committees.
Clinical research staff, including clinical research associates, coordinators, trial managers, etc.
Students aspiring to enter the clinical research industry.
Who should consider enrolling in CCRPS's groundbreaking training courses?
Whether you're already a seasoned clinical research professional or aspiring to become one, global recommendations underscore the importance of receiving GCP training and certification. Beyond being a minimum requirement, GCP certification holds several key advantages:
It serves as a formal international validation of an individual's eligibility to work in the clinical research field.
Organizations and companies rely on GCP-certified professionals to ensure compliance with industry regulations and guidelines.
GCP training imparts essential knowledge of clinical research regulations to participants.
Pharmaceutical, biotech companies, and contract research organizations prioritize hiring GCP-certified employees.
In the ever-evolving landscape of the clinical research industry in 2024, ICH GCP training has never been more crucial. Stay ahead of the curve and ensure your knowledge remains current with CCRPS's online practice training courses.
Experience the transformative impact on your clinical research career with our innovative approach to preparation and practice. Elevate your skills and stay abreast of industry developments with CCRPS today!
Download our ICH GCP Attestation Form

Good Clinical Practice GCP Training
Click this link to demo our ICH GCP training free online here!
Good Clinical Practice Training Certificate Syllabus
Introduction to Clinical Research, ICH GCP, and CFR
An Introduction to Clinical Research
An Overview of ICH GCP
Code of Federal Regulations
CFR 21 Part 11
Roles and Responsibilities (Sponsor/CRO, IRB, and Investigator)
Sponsor/CRO Responsibilities
13 Principles, IRB, & Investigator Roles
Informed Consent & Patient Safety
Adverse Event Reporting & Responsibilities
Reporting Responsibilities of the Investigators
Adverse Events
Ethical Research in Vulnerable Populations
Ethics of Research Involving Children
Ethics of Research Involving Pregnant Women and Fetuses
Ethics of Research Involving Prisoners
Trial Management, Data Handling, and Record Retention
Trial Management – Data Handling and Record Retention
Common Terminology Used In Clinical Research
Commonly Used Abbreviations and Terms in Clinical Research
ICH GCP Certification
ICH GCP Certification Exam
ICH GCP Resources
E6(R2) Good Clinical Practice: Integrated Addendum to ICH E6(R1)
FDA resource for E6 r2 addendum (also included in course)
Good Clinical Practice Resource Guide
Division of Microbiology and Infectious Diseases December 2015
WHO Library Cataloguing-in-Publication Data Handbook for good clinical research practice (GCP):
Guidance for implementation
Linkedin Resource of ICH GCP related jobs and roles
Latest trials, website updates, and more
Take courses from CCRPS and learn more on how to become a clinical research professional.
Discover more from Clinical Research Training | Certified Clinical Research Professionals Course
ICH GCP Guidelines
The ICH GCP guidelines provide public assurance that trial subjects' rights, security and well-being are protected in accord with the principles which have their source from Helsinki Declaration. Compliance ensures credible clinical data; 15 points present a unified standard for European Union (EU), Japan & United States to facilitate mutual acceptance by regulatory authorities across those jurisdictions currently compliant with WHO's good practices along side Australia Canada Nordic countries+World Health Organization
This principle has been developed with all their current good clinical practices of the European Union, Japan and USA in addition to those from Australia Canada Nordic countries World Health Organization (WHO). The guidelines established within this document may also be applied during other types medical trials which could have an effect on individual subjects' safety or well being.
ICH GCP Training Free
Here are some ICH GCP training free online guidelines. Make a quizlet or copy the ich gcp guidelines quizlet, then manually rewrite each of these into an individual card that you can easily remember because it's all on one page! To review them more effectively – combine any two cards together if they both pertain to something related in theme (examples: "Committee" & “Implementation”). Continue condensing words and combining sections until down from 50-100 flashcards; after doing so take time out every day this week before your exam(s) for practice reviewing what has been learned thus far by going through each set again slowly but thoroughly while listening carefully
Now you can get internationally accredited ICH GCP certification for $50 through CCRPS course which includes several examples in each video to solidify your knowledge.
ICH GCP Guidelines
The ICH GCP guidelines, including ich gcp e6, provides public assurance that the rights, security and also well-being of trial subjects are protected in accord with the principles which have their source in the Declaration of Helsinki. In addition, compliance ensures credible clinical trial data. The 15 ICH GCP principles presents a unified standard for the European Union (EU), Japan and the United States to facilitate the mutual acceptance of clinical data by the regulatory authorities in those jurisdictions. The principle has been developed with all their current good clinical practices of the European Union, Japan, and the USA, in Addition to those of Australia, Canada, the Nordic countries and the World Health Organization (WHO). This principle ought to be followed when generating. The principles established in this guideline may also be applied to other clinical investigations which might have an influence on the security and well-being of individual subjects.
ICH GCP Training Free
In order to self-learn ich gcp training free online:
1) make a quizlet account (or use the ich gcp guidelines quizlet)
2) manually rewrite each of the guidelines below into quizlet (this is ESSENTIAL in getting the guidelines to stick in your brain!)
3) continue condensing the words and combining guidelines until you’re down to 50-100 flashcards
4) review set 2-3 times and delete cards to clearly remember
5) continue to review and delete cards until you have it memorized!
While our ICH GCP training course Is only $50 it is essential to learning to applying ich gcp guidelines in an advanced method, you should be able to remotely memorize the guidelines on your own for free as an expert adult learner.
ICH GCP GLOSSARY
While our ICH GCP training course is essential to learning to applying ich gcp guidelines in an advanced method, you should be able to remotely memorize the guidelines on your own for free as an expert adult learner.
1. ICH GCP GLOSSARY
1.1 Adverse Drug Reaction (ADR)
From the pre-approval clinical experience with a new medicinal product or its new usages, particularly as the therapeutic dose(s) could not have been established: all noxious and unintended responses to a medicinal product related to any dose ought to be considered adverse drug reactions. The term responses to a medicinal product means that a causal relationship between a medicinal product and an adverse event is at least a reasonable chance, i.e. the connection can't be ruled out. Regarding marketed medicinal products: a reaction to a drug that is noxious and unintended and that occurs at doses normally utilized in man for prophylaxis, diagnosis, or treatment of diseases or for modification of physiological function (see the ICH Guideline for Clinical Safety Data Management: Definitions and Standards for Expedited Reporting).
1.4 Applicable Regulatory Requirement(s)
Any regulation (s) and law (s) addressing the conduct of clinical trials of investigational products.
1.5 Approval (in relation to Institutional Review Boards)
The affirmative decision of the IRB that the clinical trial was reviewed and could be conducted at the institution site within the constraints set forth by the IRB, the institution, Good Clinical Practice (GCP), and the relevant regulatory requirements.
1.7 Audit Certificate
A statement of confirmation by the auditor that an audit has happened. 1.9 Audit Trail
Documentation which allows reconstruction of the class of occasions. 1.10 Blinding/Masking
A process where a couple of parties into this trial are kept unaware of the treatment assignment(s). Single-blinding usually indicates the topic (s) being unaware, and also double-blinding usually indicates the topic (s), investigator(s), track, and, sometimes, data analyst(s) being unaware of the treatment assignment(s).
1.11 Case Report Form (CRF)
A printed, optical, or electronic document designed to record all the protocol required data to be recorded to the sponsor on each trial field.
1.12 Clinical Trial/Study
Any investigation in human subjects meant to discover or verify the clinical, psychiatric or other pharmacodynamic effects of nvestigational product(s), or to identify any adverse reactions to an investigational product(s), or to research absorption, distribution, metabolism, and excretion of an investigational product(s) together with the goal of ascertaining its security and/or effectiveness.
1.13 Clinical Trial/Study Report
A written outline of some trial/study of any therapeutic, prophylactic, or diagnostic agent conducted in human subjects, where the clinical and statistical description, presentations, and analyses are fully integrated into one report (see the ICH Guideline for Structure and Content of Clinical Study Reports).
1.14 Comparator (Product)
An investigational or marketed product (i.e., active control), or placebo, used as a benchmark in a clinical investigation.
1.15 Compliance (in relation to trials)
Adherence to all of the trial-related needs, Good Clinical Practice (GCP) requirements, and the relevant regulatory requirements.
1.17 Deal
A written, dated, and signed agreement between two or more involved parties that sets out any arrangements on delegation and distribution of tasks and duties , if appropriate, on financial issues. The protocol could serve as the foundation of a contract.
1.18 Coordinating Committee
A committee that a sponsor may organize to coordinate with the behavior of a multicentre trial.
1.19 Coordinating Investigator
An employee assigned the responsibility of the coordination of investigators at several centers participating in a multicentre trial.
1.20 Contract Research Organization (CRO)
A individual or a business (commercial, academic, or other) contracted by the sponsor to do at least one of a host's trial-related responsibilities and purposes.
1.21 Immediate Access
Permission to examine, analyze, verify, and reproduce any records and reports which are important to analysis of a medical trial. Any celebration (e.g., national and international regulatory authorities, sponsor's monitors and auditors) with direct access should take all reasonable measures within the constraints of the applicable regulatory requirement(s) to keep the confidentiality of subjects' identities and sponsor's proprietary information.
1.22 Documentation
All documents, in any kind (such as, but not restricted to, written, digital, magnetic, and optical records, and tests, x-rays, and electrocardiograms) that describe or record the methods, behavior, or effects of a trial, and the factors affecting a trial, and the action taken.
1.23 Critical Documents
Documents that individually and collectively permit evaluation of the behavior of a study and the quality of the data generated.
1.24 Good Clinical Practice (GCP)
A benchmark for the design, conduct, performance, monitoring, auditing, recording, analyses, and reporting of clinical trials that offers assurance that the data and reported results are credible and accurate, and the rights, ethics, and confidentiality of trial subjects are protected.
1.25 Independent Data-Monitoring Committee (IDMC/Data and Safety Monitoring Board, Monitoring Committee, Data Monitoring Committee)
A separate data-monitoring committee which could be determined by the sponsor to assess at intervals the progress of a clinical trial, the safety information, and the critical efficacy endpoints, and to recommend to the sponsor whether to continue, change, or discontinue a trial.
1.26 Impartial Witness
A person, who's independent of this trial, that can't be unfairly influenced by people associated in this trial, who attends the informed consent process if the subject or the subject's legally acceptable representative can't read, and who reads the informed consent form and any other written information provided to the topic.
1.27 Independent Ethics Committee (IEC)
An independent body (a review board or a committee, institutional, regional, national, or supranational), constituted of caregivers and non-medical associates, whose duty is to make sure the security of their rights, security and well-being of human issues involved in an investigation and to provide public assurance of the protection, by, among other things, reviewing and approving / providing appropriate view on, the trial procedure, the arrangement of the investigator(s), facilities, and the processes and material to be utilized in obtaining and documenting informed consent of the trial subjects.
1.28 Informed Consent
A procedure in which a subject voluntarily confirms his or her willingness to take part in a specific trial, after being informed of all details of the trial that relate to the subject of choice to engage. Informed consent is documented by way of a written, signed and dated informed consent form.
1.29 Inspection
The action by a regulatory authority(ies) of conducting an official review of documents, records, facilities, and some other sources which are deemed by the authority(ies) to be associated with the clinical trial which could be found in the website of this trial, in the host's or contract study organization's (CRO's) facilities, or at other establishments deemed appropriate by the regulatory authority(ies).
1.30 Institution (medical)
Any private or public entity or agency or medical or dental facility where clinical trials have been conducted.
1.31 Institutional Review Board (IRB)
An independent body constituted of medical, scientific, and non-scientific associates, whose duty is to guarantee the security of their rights, security and well-being of human subjects involved in a trial by, among other things, reviewing, approving, and providing continuing review of trial protocol and amendments and of the methods and substance to be utilized in obtaining and documenting informed consent of the trial subjects.
1.33 Investigational Merchandise
A pharmaceutical form of an active ingredient or placebo being tested or used as a benchmark in a clinical trial, such as a product with a marketing authorization when used or assembled (formulated or packaged) in a sense different from the approved form, or if used for an unapproved indication, or when used to get additional information regarding an approved use.
1.34 Partner
A individual accountable for the behavior of this clinical trial at a trial website. When a trial has been conducted by a group of people at a trial site, the investigator is the responsible leader of the group and might be known as the researcher. See also Subinvestigator.
1.35 Investigator / Institution
An expression meaning "the investigator and/or institution, where required by the applicable regulatory requirements".
1.36 Investigator's Brochure
A compilation of the clinical and nonclinical data on the investigational product(s) which is relevant to the study of the investigational product(s) in human subjects (see 7. Investigator’s Brochure).
1.37 Legally Acceptable Representative
An individual or juridical or other body authorized under applicable law to consent, on behalf of a prospective subject, to the subject's participation in the clinical trial.
1.38 Monitoring
The act of overseeing the progress of a clinical trial, and of ensuring that it is conducted, recorded, and reported in accordance with the protocol, Standard Operating Procedures (SOPs), Good Clinical Practice (GCP), and the applicable regulatory requirement(s).
1.39 Monitoring Report
A written report from the monitor to the sponsor after each site visit and/or other trial-related communication according to the sponsor’s SOPs.
1.40 Multicentre Trial
A clinical trial conducted according to a single protocol but at more than one site, and therefore, carried out by more than one investigator.
1.41 Nonclinical Study
Biomedical studies not performed on human subjects.
1.42 Opinion (in relation to Independent Ethics Committee)
The judgement and/or the advice provided by an Independent Ethics Committee (IEC). 1.43 Original Medical Record
See Source Documents (Below in 1.52).
1.44 Protocol
A document that describes the objective(s), design, methodology, statistical considerations, and organization of a trial. The protocol usually also gives the background and rationale for the trial, but these could be provided in other protocol referenced documents. Throughout the ICH GCP Guideline the term protocol refers to protocol and protocol amendments.
1.45 Protocol Amendment
A written description of a change(s) to or formal clarification of a protocol. 1.46 Quality Assurance (QA)
All those planned and systematic actions that are established to ensure that the trial is performed and the data are generated, documented (recorded), and reported in compliance with Good Clinical Practice (GCP) and the applicable regulatory requirement(s).
1.47 Quality Control (QC)
The operational techniques and activities undertaken within the quality assurance system to verify that the requirements for quality of the trial-related activities have been fulfilled.
1.48 Randomization
The process of assigning trial subjects to treatment or control groups using an element of chance to determine the assignments in order to reduce bias.
1.49 Regulatory Authorities
Bodies with the power to regulate. In the ICH GCP guideline the expression Regulatory Authorities includes the authorities that review submitted clinical data and those that conduct inspections (see 1.29). These bodies are sometimes referred to as competent authorities.
1.50 Serious Adverse Event (SAE) or Serious Adverse Drug Reaction (Serious ADR)
Any untoward medical occurrence that at any dose: - results in death, - is life-threatening, - requires inpatient hospitalization or prolongation of existing hospitalization, - results in persistent or significant disability/incapacity, or - is a congenital anomaly/birth defect (see the ICH Guideline for Clinical Safety Data Management: Definitions and Standards for Expedited Reporting).
1.51 Source Data
All information in original records and certified copies of original records of clinical findings, observations, or other activities in a clinical trial necessary for the reconstruction and evaluation of the trial. Source data are contained in source documents (original records or certified copies).
1.52 Source Documents
Original documents, data, and records (e.g., hospital records, clinical and office charts, laboratory notes, memoranda, subjects' diaries or evaluation checklists, pharmacy dispensing records, recorded data from automated instruments, copies or transcriptions certified after verification as being accurate copies, microfiches, photographic negatives, microfilm or magnetic media, x-rays, subject files, and records kept at the pharmacy, at the laboratories and at medico-technical departments involved in the clinical trial).
1.53 Sponsor
An individual, company, institution, or organization which takes responsibility for the initiation, management, and/or financing of a clinical trial.
1.54 Sponsor-Investigator
An individual who both initiates and conducts, alone or with others, a clinical trial, and under whose immediate direction the investigational product is administered to, dispensed to, or used by a subject. The term does not include any person other than an individual (e.g., it does not include a corporation or an agency). The obligations of a sponsor-investigator include both those of a sponsor and those of an investigator.
1.55 Standard Operating Procedures (SOPs)
Detailed, written instructions to achieve uniformity of the performance of a specific function.
1.56 Subinvestigator Any individual member of the clinical trial team designated and supervised by the investigator at a trial site to perform critical trial-related procedures and/or to make important trial-related decisions (e.g., associates, residents, research fellows). See also Investigator.
1.57 Subject/Trial Subject
An individual who participates in a clinical trial, either as a recipient of the investigational product(s) or as a control.
1.58 Subject Identification Code
A unique identifier assigned by the investigator to each trial subject to protect the subject's identity and used in lieu of the subject's name when the investigator reports adverse events and/or other trial related data.
1.59 Trial Site
The location(s) where trial-related activities are actually conducted. 1.60 Unexpected Adverse Drug Reaction
An adverse reaction, the nature or severity of which is not consistent with the applicable product information (e.g., Investigator's Brochure for an unapproved investigational product or package insert/summary of product characteristics for an approved product) (see the ICH Guideline for Clinical Safety Data Management: Definitions and Standards for Expedited Reporting).
1.61 Vulnerable Subjects
Individuals whose willingness to volunteer in a clinical trial may be unduly influenced by the expectation, whether justified or not, of benefits associated with participation, or of a retaliatory response from senior members of a hierarchy in case of refusal to participate. Examples are members of a group with a hierarchical structure, such as medical, pharmacy, dental, and nursing students, subordinate hospital and laboratory personnel, employees of the pharmaceutical industry, members of the armed forces, and persons kept in detention. Other vulnerable subjects include patients with incurable diseases, persons in nursing homes, unemployed or impoverished persons, patients in emergency situations, ethnic minority groups, homeless persons, nomads, refugees, minors, and those incapable of giving consent.
1.62 Well-being (of the trial subjects)
The physical and mental integrity of the subjects participating in a clinical trial.
2. THE PRINCIPLES OF ICH GCP
2.1 Clinical trials should be conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki, and that are consistent with GCP and the applicable regulatory requirement(s).
2.2 Before a trial is initiated, foreseeable risks and inconveniences should be weighed against the anticipated benefit for the individual trial subject and society. A trial should be initiated and continued only if the anticipated benefits justify the risks.
2.3 The rights, safety, and well-being of the trial subjects are the most important considerations and should prevail over interests of science and society.
2.4 The available nonclinical and clinical information on an investigational product should be adequate to support the proposed clinical trial.
2.5 Clinical trials should be scientifically sound, and described in a clear, detailed protocol.
2.6 A trial should be conducted in compliance with the protocol that has received prior institutional review board (IRB)/independent ethics committee (IEC) approval/favourable opinion.
2.7 The medical care given to, and medical decisions made on behalf of, subjects should always be the responsibility of a qualified physician or, when appropriate, of a qualified dentist.
2.8 Each individual involved in conducting a trial should be qualified by education, training, and experience to perform his or her respective task(s).
2.9 Freely given informed consent should be obtained from every subject prior to clinical trial participation.
2.10 All clinical trial information should be recorded, handled, and stored in a way that allows its accurate reporting, interpretation and verification.
2.11 The confidentiality of records that could identify subjects should be protected, respecting the privacy and confidentiality rules in accordance with the applicable regulatory requirement(s).
2.12 Investigational products should be manufactured, handled, and stored in accordance with applicable good manufacturing practice (GMP). They should be used in accordance with the approved protocol.
2.13 Systems with procedures that assure the quality of every aspect of the trial should be implemented.
3. INSTITUTIONAL REVIEW BOARD/INDEPENDENT ETHICS COMMITTEE (IRB/IEC)
3.1 Responsibilities
3.1.1 An IRB/IEC should safeguard the rights, safety, and well-being of all trial subjects. Special attention should be paid to trials that may include vulnerable subjects.
3.1.2 The IRB/IEC should obtain the following documents: trial protocol(s)/amendment(s), written informed consent form(s) and consent form updates that the investigator proposes for use in the trial, subject recruitment procedures (e.g. advertisements), written information to be provided to subjects, Investigator's Brochure (IB), available safety information, information about payments and compensation available to subjects, the investigator’s current curriculum vitae and/or other documentation evidencing qualifications, and any other documents that the IRB/IEC may need to fulfil its responsibilities. The IRB/IEC should review a proposed clinical trial within a reasonable time and document its views in writing, clearly identifying the trial, the documents reviewed and the dates for the following: - approval/favourable opinion; - modifications required prior to its approval/favourable opinion; - disapproval / negative opinion; and - termination/suspension of any prior approval/favourable opinion.
3.1.3 The IRB/IEC should consider the qualifications of the investigator for the proposed trial, as documented by a current curriculum vitae and/or by any other relevant documentation the IRB/IEC requests.
3.1.4 The IRB/IEC should conduct continuing review of each ongoing trial at intervals appropriate to the degree of risk to human subjects, but at least once per year. 3.1.5 The IRB/IEC may request more information than is outlined in paragraph 4.8.10 be given to subjects when, in the judgement of the IRB/IEC, the additional information would add meaningfully to the protection of the rights, safety and/or well-being of the subjects.
3.1.6 When a non-therapeutic trial is to be carried out with the consent of the subject’s legally acceptable representative (see 4.8.12, 4.8.14), the IRB/IEC should determine that the proposed protocol and/or other document(s) adequately addresses relevant ethical concerns and meets applicable regulatory requirements for such trials.
3.1.7 Where the protocol indicates that prior consent of the trial subject or the subject’s legally acceptable representative is not possible (see 4.8.15), the IRB/IEC should determine that the proposed protocol and/or other document(s) adequately addresses relevant ethical concerns and meets applicable regulatory requirements for such trials (i.e. in emergency situations).
3.1.8 The IRB/IEC should review both the amount and method of payment to subjects to assure that neither presents problems of coercion or undue influence on the trial subjects. Payments to a subject should be prorated and not wholly contingent on completion of the trial by the subject.
3.1.9 The IRB/IEC should ensure that information regarding payment to subjects, including the methods, amounts, and schedule of payment to trial subjects, is set forth in the written informed consent form and any other written information to be provided to subjects. The way payment will be prorated should be specified.
3.2 Composition, Functions and Operations
3.2.1 The IRB/IEC should consist of a reasonable number of members, who collectively have the qualifications and experience to review and evaluate the science, medical aspects, and ethics of the proposed trial. It is recommended that the IRB/IEC should include: (a) At least five members. (b) At least one member whose primary area of interest is in a nonscientific area. (c) At least one member who is independent of the institution/trial site. Only those IRB/IEC members who are independent of the investigator and the sponsor of the trial should vote/provide opinion on a trial-related matter. A list of IRB/IEC members and their qualifications should be maintained.
3.2.2 The IRB/IEC should perform its functions according to written operating procedures, should maintain written records of its activities and minutes of its meetings, and should comply with GCP and with the applicable regulatory requirement(s).
3.2.3 An IRB/IEC should make its decisions at announced meetings at which at least a quorum, as stipulated in its written operating procedures, is present.
3.2.4 Only members who participate in the IRB/IEC review and discussion should vote/provide their opinion and/or advise.
3.2.5 The investigator may provide information on any aspect of the trial, but should not participate in the deliberations of the IRB/IEC or in the vote/opinion of the IRB/IEC.
3.2.6 An IRB/IEC may invite nonmembers with expertise in special areas for assistance.
3.3 Procedures
The IRB/IEC should establish, document in writing, and follow its procedures, which should include:
3.3.1 Determining its composition (names and qualifications of the members) and the authority under which it is established.
3.3.2 Scheduling, notifying its members of, and conducting its meetings. 3.3.3 Conducting initial and continuing review of trials.
3.3.4 Determining the frequency of continuing review, as appropriate.
3.3.5 Providing, according to the applicable regulatory requirements, expedited review and approval/favourable opinion of minor change(s) in ongoing trials that have the approval/favourable opinion of the IRB/IEC.
3.3.6 Specifying that no subject should be admitted to a trial before the IRB/IEC issues its written approval/favourable opinion of the trial.
3.3.7 Specifying that no deviations from, or changes of, the protocol should be initiated without prior written IRB/IEC approval/favourable opinion of an appropriate amendment, except when necessary to eliminate immediate hazards to the subjects or when the change(s) involves only logistical or administrative aspects of the trial (e.g., change of monitor(s), telephone number(s)) (see 4.5.2).
3.3.8 Specifying that the investigator should promptly report to the IRB/IEC: (a) Deviations from, or changes of, the protocol to eliminate immediate hazards to the trial subjects (see 3.3.7, 4.5.2, 4.5.4). (b) Changes increasing the risk to subjects and/or affecting significantly the conduct of the trial (see 4.10.2). (c) All adverse drug reactions (ADRs) that are both serious and unexpected. (d) New information that may affect adversely the safety of the subjects or the conduct of the trial.
3.3.9 Ensuring that the IRB/IEC promptly notify in writing the investigator/institution concerning: (a) Its trial-related decisions/opinions. (b) The reasons for its decisions/opinions. (c) Procedures for appeal
ICH GCP
GCP Online Course: Advanced ICH GCP
Certification (AGCPC)
ENROLL
What Is ICH GCP Certification
Home Course Catalog
Career Guides
Group Orders
Enroll
of its decisions/opinions.
of its decisions/opinions.
3.4 Records
The IRB/IEC should retain all relevant records (e.g., written procedures, membership lists, lists of occupations/affiliations of members, submitted documents, minutes of meetings, and correspondence) for a period of at least 3 years after completion of the trial and make them available upon request from the regulatory authority(ies). The IRB/IEC may be asked by investigators, sponsors or regulatory authorities to provide its written procedures and membership lists.
4. INVESTIGATOR
4.1 Investigator's Qualifications and Agreements
4.1.1 The investigator(s) should be qualified by education, training, and experience to assume responsibility for the proper conduct of the trial, should meet all the qualifications specified by the applicable regulatory requirement(s), and should provide evidence of such qualifications through up-to-date curriculum vitae and/or other relevant documentation requested by the sponsor, the IRB/IEC, and/or the regulatory authority(ies).
4.1.2 The investigator should be thoroughly familiar with the appropriate use of the investigational product(s), as described in the protocol, in the current Investigator's Brochure, in the product information and in other information sources provided by the sponsor.
4.1.3 The investigator should be aware of, and should comply with, GCP and the applicable regulatory requirements.
4.1.4 The investigator/institution should permit monitoring and auditing by the sponsor, and inspection by the appropriate regulatory authority(ies).
4.1.5 The investigator should maintain a list of appropriately qualified persons to whom the investigator has delegated significant trial-related duties.
4.2 Adequate Resources
4.2.1 The investigator should be able to demonstrate (e.g., based on retrospective data) a potential for recruiting the required number of suitable subjects within the agreed recruitment period.
4.2.2 The investigator should have sufficient time to properly conduct and complete the trial within the agreed trial period.
4.2.3 The investigator should have available an adequate number of qualified staff and adequate facilities for the foreseen duration of the trial to conduct the trial properly and safely.
4.2.4 The investigator should ensure that all persons assisting with the trial are adequately informed about the protocol, the investigational product(s), and their trial-related duties and functions.
4.3 Medical Care of Trial Subjects
4.3.1 A qualified physician (or dentist, when appropriate), who is an investigator or a sub-investigator for the trial, should be responsible for all trial-related medical (or dental) decisions.
4.3.2 During and following a subject's participation in a trial, the investigator/institution should ensure that adequate medical care is provided to a subject for any adverse events, including clinically significant laboratory values, related to the trial. The investigator/institution should inform a subject when medical care is needed for intercurrent illness(es) of which the investigator becomes aware.
4.3.3 It is recommended that the investigator inform the subject's primary physician about the subject's participation in the trial if the subject has a primary physician and if the subject agrees to the primary physician being informed.
4.3.4 Although a subject is not obliged to give his/her reason(s) for withdrawing prematurely from a trial, the investigator should make a reasonable effort to ascertain the reason(s), while fully respecting the subject's rights.
4.4 Communication with IRB/IEC
4.4.1 Before initiating a trial, the investigator/institution should have written and dated approval/favourable opinion from the IRB/IEC for the trial protocol, written informed consent form, consent form updates, subject recruitment procedures (e.g., advertisements), and any other written information to be provided to subjects.
4.4.2 As part of the investigator's/institution’s written application to the IRB/IEC, the investigator/institution should provide the IRB/IEC with a current copy of the Investigator's Brochure. If the Investigator's Brochure is updated during the trial, the investigator/institution should supply a copy of the updated Investigator’s Brochure to the IRB/IEC.
4.4.3 During the trial the investigator/institution should provide to the IRB/IEC all documents subject to review.
4.5 Compliance with Protocol
4.5.1 The investigator/institution should conduct the trial in compliance with the protocol agreed to by the sponsor and, if required, by the regulatory authority(ies) and which was given approval/favourable opinion by the IRB/IEC.
The investigator/institution and the sponsor must sign the protocol, or another contract, to confirm arrangement.
4.5.2 The investigator shouldn't implement any deviation from, or modifications of this protocol without agreement by the sponsor and prior review and documented approval/favourable opinion in the IRB/IEC of an amendment, except where necessary to eliminate an immediate hazard(s) to trial subjects, or when the change(s) involves only logistical or administrative aspects of the trial (e.g., alter in track (s), change of phone number(s)).
4.5.4 The investigator may implement a deviation from, or a reversal of, the protocol to eliminate an immediate hazard(s) to trial subjects without previous IRB/IEC approval/favourable opinion. When possible, the implemented deviation or change, the causes of this, also, if appropriate, the proposed protocol amendment(s) ought to be filed: (a) into the IRB/IEC for inspection and approval/favourable view, (b) to the sponsor for agreement and, when necessary, (c) into the regulatory authority(ies).
4.6.2 Where allowed/required, the investigator/institution may/should assign some or all the investigator's/institution's duties for investigational product(s) accountability at the trial site(s ) ) to an proper pharmacist or another suitable person who's under the oversight of their investigator/institution. .
4.6.3 The investigator/institution or a pharmacist or other appropriate person, who's given by the investigator/institution, should keep records of their item's delivery to the trial site, the inventory at the website, the usage by each topic, and also the yield to the sponsor or alternative disposition of unused product(s). These records must include dates, numbers, batch/serial numbers, expiration dates (if applicable), and the exceptional code numbers assigned to the investigational product(s) and trial subjects. Researchers should keep records that document adequately that the subjects were provided the doses specified by the protocol and reconcile all investigational product(s) obtained from the host.
4.6.4 The investigational product(s) ought to be kept as defined by the host (see 5.13.2 and 5.14.3) and in compliance with applicable regulatory requirement(s).
4.6.5 The investigator should ensure that the investigational product(s) are utilized only in compliance with the accepted protocol.
4.6.6 The investigator, or a person designated by the investigator/institution, must describe the proper use of the investigational product(s) to each subject and should check, at times appropriate for the trial, that each subject is following the directions correctly. If the trial is blinded, the investigator should promptly document and explain to the sponsor any early unblinding (e.g., accidental unblinding, unblinding because of a serious adverse event) of the investigational product(s).
4.8 Informed Consent of Trial Subjects
4.8.1 In obtaining and documenting informed consent, the investigator must comply with the applicable regulatory requirement(s), and should adhere to GCP and to the ethical principles which have their source in the Declaration of Helsinki. Before the start of the trial, the investigator needs to have the IRB/IEC's composed approval/favourable view of this written informed consent form and any other written information to be offered to subjects.
4.8.2 The written informed consent form and any other written information to be given to subjects must be revised whenever important new information becomes available that may be pertinent to this subject's approval. Any revised written informed consent form, and written advice must get the IRB/IEC's approval/favourable view ahead of usage. The subject or the subject's legally acceptable representative ought to be informed in a timely fashion if new information becomes available that may be pertinent to the subject's willingness to continue participation in the trial. The communication of the information ought to be documented.
4.8.5 The investigator, or a person designated by the investigator, should fully inform the subject or, even if the topic is not able to give informed consent, the subject's legally acceptable representative, of all pertinent aspects of the trial including the written advice and also the approval/ favourable opinion by the IRB/IEC.
4.8.6 The language used in the written and oral information regarding the trial, including the written informed consent form, must be non-technical as functional and should be clear to the subject or the subject's legally acceptable representative and the impartial witness, where applicable.
4.8.7 Before informed consent may be obtained, the investigator, or someone designated by the investigator, should offer the subject or the subject's legally acceptable representative ample time and opportunity to ask about details of the trial and also to choose whether or not to take part in the trial. All queries concerning the trial ought to be answered to the satisfaction of the topic or the subject's legally acceptable representative.
4.8.8 Before a subject's involvement in the analysis, the written informed consent form ought to be signed and dated by the topic or from the subject's legally appropriate agent, and from the man who conducted the informed consent discussion.
4.8.9 If a topic can't read or if a legally acceptable representative is not able to read, an impartial witness should be present throughout the entire informed consent discussion. Following the written informed consent form and any other written information to be given to subjects, will be read and explained to the subject or the subject's legally acceptable representative, and after the subject or the subject's legally acceptable representative has orally consented to the subject of involvement in the trial and, if capable of doing this, has signed and dated the informed consent form, the witness must sign and personally date the consent form. (b) The intention of the trial. If there's not any intended clinical benefit to the subject, the topic ought to be made aware of the (I) The alternative procedure(s) or course(s) of treatment which could be accessible to the matter, and their important potential benefits and hazards. (j) The reimbursement and/or therapy readily available to the subject at case of trial-related injury. (l) The anticipated expenses, if any, to the subject of participating in the trial. (n) The monitor(s), the auditor(s), the IRB/IEC, along with the regulatory authority(ies) will be granted direct entry to the subject's original medical records for verification of clinical trial processes and/or information, without violating the confidentiality of this topic, to the extent allowed by the applicable legislation and regulations and that, by signing a written informed consent form, the subject or the subject's legally acceptable representative is authorizing such access. (o) That records identifying the subject will be kept confidential and, to the extent allowed by regulations or laws, won't be made publicly accessible. If the outcomes of the trial have been published, the subject's identity will stay confidential. (p) The subject or the subject's legally acceptable representative will be informed in a timely manner if information becomes available that may be pertinent to the subject's willingness to continue participation in the trial. (q) The individual (s) to contact for more information concerning the trial and the rights of trial subjects, and whom to contact in case of trial-related injury. (r) The foreseeable conditions and/or motives under the subject's involvement in the trial could be terminated. (s) The expected duration of the subject's involvement in the trial. (t) The approximate number of subjects included with the trial.
4.8.11 Prior to participation in the trial, the subject or the subject's legally acceptable representative should be given a copy of the signed and dated written informed consent form and any other written information supplied to the topics. During a subject's involvement in the trial, the subject or the subject's legally acceptable representative should get a copy of the signed and dated consent form updates and a copy of any amendments to the written information given to subjects.
4.8.12 When a clinical trial (therapeutic or non-therapeutic) includes subjects who can only be registered in the trial with the permission of the subject's legally acceptable representative (e.g., minors, or individuals with severe dementia), the subject ought to be informed about the trial to the extent compatible with the subject of comprehension and, if capable, the subject should sign and personally date the written informed consent.
4.8.13 Except as described in 4.8.14, a non-therapeutic trial (i.e. a trial where there is not any anticipated direct clinical benefit to the subject), needs to be conducted in subjects who give consent and that sign and date the written informed consent form.
4.8.14 Non-therapeutic trials might be conducted in subjects with consent of a legally acceptable representative provided that the following requirements are fulfilled: (a) The aims of the trial can't be fulfilled by way of a trial in subjects that can provide informed consent. (c) The adverse influence on the subject's well-being is reduced and minimized. (d) The trial isn't prohibited by legislation. (e) The approval/favorable view of this IRB/IEC is especially sought on the inclusion of these topics, and the written approval/ favorable opinion covers this aspect. Topics in such trials must be particularly closely monitored and must be removed if they seem to be overly distressed.
4.8.15 In crisis situations, when prior permission of the subject isn't feasible, the permission of the subject's legally acceptable representative, if present, should be asked. When previous consent of the topic isn't feasible, along with the subject's legally acceptable representative isn't available, enrollment of this topic ought to require steps described in the protocol or elsewhere, with recorded approval/favorable opinion from the IRB/IEC, to safeguard the rights, security and well-being of this topic and also to guarantee compliance with applicable regulatory requirements. The subject or the subject's legally acceptable representative ought to be informed about the trial when possible and agree to continue along with additional approval as appropriate (see 4.8.10) ought to be asked.
4.9 Records and Reports
4.9.1 The investigator should ensure the accuracy, completeness, legibility, and timeliness of their information reported to the host at the CRFs and in all necessary reports.
4.9.2 Data reported on the CRF, which are derived from source documents, ought to be in accordance with the source documents or the discrepancies should be clarified.
4.9.3 Any alteration or correction to a CRF ought to be dated, initialed, and explained (if necessary) and shouldn't obscure the original entry (i.e. an audit trail ought to be preserved ); that applies to both written and electronic changes or corrections (visit 5.18.4 (n)). Sponsors should provide advice to investigators or the researchers' designated representatives on making such corrections. Sponsors must have written procedures to ensure corrections or changes in CRFs created by sponsor's designated agents are recorded, are needed, and are backed by the investigator. The investigator must maintain records of the corrections and changes.
4.9.4 The investigator/institution must keep the trial documents as stated in Essential Documents for the Conduct of a Clinical Trial (see 8.) The investigator/institution must take steps to avoid accidental or premature destruction of those records.
4.9.5 Essential documents should be retained until at least two years following the final approval of a marketing application in an ICH region and until there are no pending or contemplated marketing applications in an ICH region or at least two years have elapsed since the formal discontinuation of clinical development of the investigational item. These records should be kept for a longer period however if required by the relevant regulatory requirements or by having an arrangement with the host. It's the obligation of the host to notify the investigator/institution concerning when these documents no longer have to be kept (see 5.5.12).
4.9.6 The financial details of the trial ought to be recorded in an agreement between the host and the investigator/institution.
4.9.7 Upon request of the monitor, auditor, IRB/IEC, or regulatory authority, the investigator/institution must make readily available for direct access all requested trial-related documents.
4.10 Progress Reports
4.10.1 The investigator must submit written summaries of this trial status to the IRB/IEC yearly, or more often, if asked by the IRB/IEC.
4.10.2 The investigator should promptly provide written reports on the host, the IRB/IEC (see 3.3.8) and, where applicable, the institution on any changes significantly affecting the behavior of this trial, or raising the risk to subjects. The follow-up and immediate reports must identify subjects by unique code numbers assigned to the trial subjects instead of from the subjects' names, personal identification numbers, or addresses. The investigator must also comply with the applicable regulatory requirement(s) associated with the reporting of unexpected serious adverse drug reactions to the regulatory authority(ies) along with the IRB/IEC.
4.11.2 Adverse laboratory or events abnormalities identified in the protocol as critical to safety evaluations should be reported to the host in line with the coverage requirements and within the time intervals specified by the host in the protocol.
4.12 Premature Termination or Suspension of a Trial
In case the trial is prematurely terminated or suspended for any reason, the investigator/institution should immediately inform the trial issues, should guarantee proper treatment and follow-up for those issues, and, where required by the applicable regulatory requirement(s), should notify the regulatory authority(ies). Additionally:
4.12.1 If the investigator terminates or suspends a trial without prior agreement of the host, the investigator must inform the institution where applicable, and also the investigator/institution should immediately notify the host and the IRB/IEC, and should offer the sponsor along with the IRB/IEC a detailed written explanation of the termination or suspension.
4.12.2 If the sponsor terminates or suspends a trial (see 5.21), the investigator must immediately notify the institution where applicable along with the investigator/institution should immediately inform the IRB/IEC and supply the IRB/IEC a detailed written explanation of the termination or suspension.
4.12.3 When the IRB/IEC terminates or suspends its approval/favorable view of a trial (see 3.1.2 and 3.3.9), the investigator must inform the institution where applicable along with the investigator/institution should immediately notify the host and supply the sponsor with a detailed written explanation of the termination or suspension.
4.13 Final Report(s) by Investigator
Upon completion of the trial, the investigator, where relevant, should notify the institution; the investigator/institution must offer the IRB/IEC using a review of the trial's result, and the regulatory authority(ies) with any reports required.
5. SPONSOR
5.1 Quality Assurance and Quality Control
5.1.1 The host is responsible for implementing and maintaining quality assurance and quality management systems with written SOPs to make certain that trials are conducted and data are generated, documented (recorded), and documented according to the protocol, GCP, and the applicable regulatory requirement(s).
5.1.2 The host is responsible for securing agreement from all involved parties to ensure immediate access (see 1.21) to each of trial related websites, origin data/documents, and reports for the purpose of monitoring and auditing by the sponsor, and review by domestic and international regulatory authorities.
5.1.3 Quality control ought to be applied to every point of data handling to make sure that all data are reliable and have been processed properly.
5.1.4 Agreements, created by the host with all the investigator/institution and some other parties involved with the clinical trial, must be in writing, within this protocol or in another arrangement.
5.2 Contract Research Organization (CRO)
5.2.1 A sponsor may transfer any or all the host's trial-related responsibilities and functions to a CRO, but the ultimate responsibility for its integrity and quality of the trial data always resides with the host. The CRO should apply quality assurance and quality management.
5.2.2 Any trial-related responsibility and function that's transferred to and assumed by a CRO ought to be given in writing.
5.2.3 Any trial-related responsibilities and functions not specifically transferred to and assumed by a CRO are retained by the host.
5.2.4 All references to a host within this guideline apply to a CRO to the extent that a CRO has assumed the trial related duties and functions of a host.
5.3 Medical Experience
The sponsor must designate suitably qualified medical staff that are easily available to counsel trial related health questions or issues. If needed, external advisor (s) can be made for this function.
5.4 Trial Design
5.4.1 The host must use qualified people (e.g. biostatisticians, clinical pharmacologists, and physicians) as appropriate, during all phases of their trial process, in designing the protocol and CRFs and preparing the investigations into assessing and preparing interim and final clinical trial reports.
5.5 Trial Management, Data Handling, and Record Keeping i.e. ICH GCP guidelines for clinical data management
5.5.1 The host must use appropriately qualified people to supervise the general conduct of this trial, to deal with the information, to confirm the information, to conduct the statistical analyses, and also to prepare the demo reports. The IDMC should have written operating procedures and keep written records of its meetings.
5.5.3 When using electronic trial data management and/or remote electronic trial information programs, the host needs to: (a) Ensure and document that the electronic data processing procedure(s) adheres to the sponsor's established requirements for completeness, accuracy and reliability, and consistent intended performance (i.e. identification ). (b) Maintains SOPs for utilizing such systems. (c) Make sure that these systems are intended to allow data changes in such a manner in which the data changes are documented and that there isn't any deletion of input data (i.e. keep an audit trail, information path, edit path ). (d) Keep a safety system which prevents unauthorized access into this information. (e) Keep a listing of those people that are licensed to produce information modifications (see 4.1.5 and 4.9.3). (g) Shield the blinding, if some (e.g. keep the data during data entry and processing system ).
5.5.4 When data are transformed during processing, then it must remain possible to evaluate the initial observations and data with the data that is processed.
5.5.5 The host must utilize an unambiguous subject identification code (visit 1.58) which enables identification of all of the information reported for every topic.
5.5.6 The host, along with other owners of all this information, should keep each the sponsor-specific necessary documents of interest to the trial (see 8).
5.5.7 The sponsor must maintain all sponsor-specific necessary files in conformance with all the applicable regulatory requirement(s) of this country(ies) in which the item is accepted, or at which the sponsor intends to submit an application for approval(s).
5.5.9 If the sponsor discontinues the clinical development of an investigational solution, the sponsor must notify all of the trial investigators/institutions and most of the regulatory authorities.
5.5.10 Any transfer of possession of this information must be reported on the proper authority(ies), according to the applicable regulatory requirement(s).
5.5.12 The sponsor must notify the investigator(s)/association(s) in writing of their requirement for document retention and should inform the investigator(s)/association(s) in writing whenever the trial associated documents are no more needed.
5.6 Investigator Choice
5.6.1 The host is responsible for picking the investigator(s)/association (s). Each investigator ought to be qualified by experience and training and should have sufficient funds (see 4.1, 4.2) to properly conduct the trial where the investigator is chosen. If business of some coordinating committee or choice of coordinating investigator(s) will be used in multicentre trials, their company and/or choice are the host's responsibility.
5.6.2 Before entering an agreement with an investigator/institution to perform a trial, the sponsor must offer the investigator(s)/association (s) using the routine and also an up-to-date Investigator's Brochure, and should provide adequate time for your investigator/institution to assess the protocol and also the info supplied.
5.6.3 The sponsor must obtain the investigator's/institution's arrangement: (a) to conduct the trial according to GCP, together with all the applicable regulatory requirement(s) (see 4.1.3), also with the protocol agreed to by the host and given approval/favorable remark by the IRB/IEC (see 4.5.1); (b) to comply with processes for information recording/reporting; (c) to allow tracking, auditing and review (see 4.1.4) and (d) to keep the trial associated essential files until the host informs the investigator/institution that these records are no longer required (see 4.9.4 along with also 5.5.12). The host and the investigator/institution need to sign the protocol, or an alternate file, to verify this arrangement.
5.7 Allocation of Duties
Before initiating a trial, the sponsor should define, establish, and devote most of of trial-related responsibilities and purposes.
5.8 Compensation to Subjects and Investigators
5.8.1 If required by the applicable regulatory requirement(s), the host must offer insurance or if indemnify (valid and fiscal policy ) that the investigator/the association against claims arising out of the trial, and except for claims that arise from prosecution and/or neglect.
5.8.2 The host's policies and procedures must deal with the expenses of treatment for trial issues in case of trial-related accidents in agreement with the applicable regulatory requirement(s).
5.8.3 When identification subjects receive reimbursement, the procedure and way of reimbursement must comply with applicable regulatory requirement(s).
5.9 Funding
The financial facets of the trial ought to be recorded in an agreement between the host and the investigator/institution.
5.10 Notification/Submission into Regulatory Authority(ies)
Prior to initiating the clinical investigation (s), the host (or the host and the investigator, even when necessary by the applicable regulatory requirement(s)) must submit any necessary program (s) into the proper authority(ies) for inspection, approval, and/or consent (as needed by the applicable regulatory requirement(s)) to commence the trial(s). Any notification/submission ought to be dated and include adequate information to recognize the routine. (b) A statement obtained in the IRB/IEC it is organized and functions in accordance with GCP and the applicable regulations and laws. (c) Documented IRB/IEC approval/favourable view and, when requested by the host, a recent backup of protocol, written informed consent form(s) and any other written information to be offered to areas, subject recruiting processes, and records associated with payments and reimbursement available to the topics, and some other files the IRB/IEC could have asked.
5.11.2 If the IRB/IEC states its approval/favourable view upon modification (s) in almost any feature of the trial, including alteration (s) of this protocol, written informed consent form and any other written information to be offered to areas, or other processes, the sponsor must obtain in the investigator/institution that a duplicate of the modification(s) created and the date approval/favourable remark was given from the IRB/IEC.
5.11.3 The sponsor must obtain in the investigator/institution dates and documentation of any IRB/IEC reapprovals/re-evaluations with hierarchical view, also of any withdrawals or suspensions of all approval/favourable view.
5.12 Information on Investigational Product(s) 5.12.1 When planning trials, the sponsor must ensure that adequate safety and efficacy information from nonclinical clinical or studies trials are readily available to support human vulnerability from the path, in the doses, for its length, and at the trial population to be analyzed.
5.12.2 The host must upgrade the Investigator's Brochure as important new information becomes available (see 7).
5.13 Manufacturing, Packaging, Labeling, and Coding Investigational Product(s)
5.13.1 The host should ensure that the investigational product(s) (such as active comparator(s) and placebo( if appropriate ) is distinguished as appropriate for the stage of growth of the item (s), is fabricated according to any relevant GMP, and can be coded and tagged in a way that safeguards the blinding, if appropriate. Additionally, the labelling must comply with all applicable regulatory requirement(s).
5.13.2 The sponsor must determine, for the investigational product(s), decent storage temperatures, storage requirements (e.g. protection against mild ), storage times, reconstitution fluids and processes, and apparatus for product extract, if any. The host should notify all parties that are involved (e.g. tracks, researchers, pharmacistsand storage managers) of those determinations.
5.13.3 The investigational product(s) ought to be packed to avoid contamination and improper deterioration during storage and transport.
5.13.4 In clinical trials, the programming system to its investigational product(s) must incorporate a mechanism which allows rapid identification of their item (s) if a health crisis, but doesn't permit imperceptible fractures of this blinding.
5.13.5 If significant formulation changes are produced in the investigational or comparator product(s) throughout the course of clinical improvement, the outcomes of some further studies of the formulated product(s) (e.g. stability, dissolution rate, bioavailability) required to evaluate whether these changes could significantly alter the pharmacokinetic profile of this item ought to be available before using this newest formula in clinical trials.
5.14 Supplying and Handling Investigational Product(s)
5.14.1 The host is responsible for providing the investigator(s)/association (s) using all the investigational product(s ) ).
5.14.2 The host shouldn't provide an investigator/institution using the investigational product(s) before the host obtains all necessary documentation (e.g. approval/favorable view from IRB/IEC and regulatory authority(ies)).
5.14.3 The host must ensure that written procedures contain directions the investigator/institution must follow to the storage and handling of investigational product(s) for your trial and documentation . The processes should address decent and safe receipt, handling, storage, unloading, recovery of fresh product in issues, and yield of unused investigational product(s) to the host (or other disposition if approved by the host and in accordance with all the applicable regulatory requirement(s)).
5.14.4 The host needs to: (a) guarantee timely delivery of investigational product(s) into this investigator(s). (b) Keep records that document dispatch, receipt, disposition, reunite, and also destruction of this investigational product(s) (see 8). (c) Maintain a method for regaining investigational products and recording that this recovery (e.g. for deficient product remember, recover after trial completion( expired merchandise recover ).
5.14.5 The host needs to: (a) Take action to make certain that the investigational product(s) are steady over the length of usage. (b) Maintain adequate quantities of the investigational product(s) utilized from the trials to reconfirm specifications, so should it be necessary, and keep records of batch sample investigations and attributes. To the degree equilibrium allows, samples must be kept either before the investigations of the trial data will be complete or as needed by the applicable regulatory requirement(s), whichever reflects the longer retention interval.
5.15 Record Access
5.15.1 The host must ensure it is given in the protocol or other written agreement which the investigator(s)/association (s) offer immediate access to source data/documents such as trial- related observation, Tests, IRB/IEC inspection, and regulatory review.
5.15.2 The host must verify that every subject has agreed, in writing, to guide accessibility to their own first medical records to get trial-related observation, audit, IRB/IEC inspection, and regulatory scrutiny.
5.16.2 The sponsor must immediately notify all concerned investigator(s)/association (s) and the regulatory authority(ies) of findings which could affect negatively the security of topics, affect the behavior of this trial, or change the IRB/IEC's approval/favourable view to keep the test.
5.17.3 The sponsor must submit to the regulatory authority(ies) all security upgrades and periodic reports, and according to applicable regulatory requirement(s).
5.18 Tracking
5.18.1 Purpose
The functions of trial monitoring are to confirm: (a) The rights and also well-being of individual subjects are protected. (c) The conduct of the offense will be in accordance with the approved protocol/amendment(s), with GCP, along with all the applicable regulatory requirement(s). (b ) Monitors must be suitably trained, and ought to possess the clinical or scientific knowledge required to track the trial satisfactorily. A track's qualifications must be recorded.
5.18.3 Extent and Nature of Monitoring
The host should ensure that the trials have been adequately tracked. The sponsor must determine the right scope and nature of observation. The conclusion of the scope and nature of monitoring should be determined by factors like the purpose, function, style, complexity, blinding, size, and endpoints of this trial. Generally speaking there's a demand for onsite observation, before, during, and after the trialhowever in extraordinary circumstances the host may decide that central observation in combination with processes such as researchers' meetings and training, and comprehensive written advice can assure proper conduct of the trial in agreement with GCP. Statistically controlled sampling could be an acceptable way of selecting the information to be checked.
5.18.4 Monitor's Responsibilities
The track (s) in compliance with the host's requirements need to make sure that the trial will be conducted and recorded properly by executing the following actions when relevant and essential to this trial and the crime website: (a) Acting as the major field of communication between the host and the investigator. (b) Verifying that the investigator has sufficient qualifications and tools (see 4.1, 4.2, 5.6) and stay adequate throughout the trial period, which facilities, such as labs and equipment, and employees, are sufficient to safely and properly conduct the trial and stay adequate throughout the trial period. (ii) The investigational product(s) are provided exclusively to subjects that are qualified for it and in the protocol given dose(s). (iii) That matters are supplied with necessary education on correctly using, managing, storing, and returning to the investigational product(s). (iv) The reception, use, and yield of this investigational product(s) in the trial sites are regulated and recorded adequately. (v) The disposition of unused investigational product(s) in the trial sites complies with all applicable regulatory requirement(s) and can be in accord with the sponsor. (e) Verifying that written informed consent was obtained prior to each subject's involvement in this trial. (f) Ensuring that the investigator gets the current Investigator's brochure, all records, and all of the trial provides required to conduct the trial properly and also to comply with the applicable regulatory requirement(s). (h) Verifying that the investigator and the investigator's trial staff are currently still doing the given trial purposes, in accord with the protocol along with another written agreement between the host and the investigator/institution, also haven't assigned the functions to unauthorized people. (I) Verifying that the investigator will be enroling only qualified subjects. (j) Reporting the matter recruitment rate. (k) Verifying that source files and other trial documents are true, complete, retained up-to-date and preserved. (Id) Verifying that the investigator provides all of the essential documents, notifications, applications, and admissions, and these records are accurate, comprehensive, timely, legible, dated, and also establish that the trial. (m) Assessing the accuracy and completeness of the CRF entries, source files and other trial-related documents contrary to each other. The monitor especially should confirm that: (I) The information required by the protocol have been reported right about the CRFs and therefore are in accordance with the source files. (ii) Any dose or treatment alterations are well documented for all the trial issues. (iii) Adverse events, concomitant medications and inter-current disorders are reported with regard to the routine in the CRFs. (iv) Visits the subjects don't create, tests which aren't conducted, and tests which aren't performed are reported as such on the CRFs. (n) Informing the inheritance of any CRF entrance mistake, omission, or illegibility. The monitor must ensure that proper adjustments, additions, or deletions are made, obsolete, clarified (if needed ), and initialed by the investigator or from a part of the investigator's trial staff who's licensed to first CRF modifications to your investigator. This consent ought to be documented. (o) Deciding whether adverse events (AEs) are reported over the time intervals required by GCP, the protocol, the IRB/IEC, the host, as well as the applicable regulatory requirement(s). (de) Deciding if the investigator is keeping the vital files (see 8. )
5.18.5 Monitoring Procedures
The track (s) must occur after the host's established written SOPs in addition to those processes which are given by the host for tracking a particular trial.
5.18.6 Monitoring Report
(a) The screen must submit an official report to the host after every trial-site see or trial-related communication. (b) Reports must include the date, website, title of the track, and title of the investigator or other person (s) contacted. (c) Reports must include a summary of the track reviewed along with the track's statements regarding the substantial findings/facts, deviations and deficiencies, decisions and actions taken or to be obtained and/or activities recommended to procure compliance. (d) The follow-up and review of this observation report with all the host ought to be recorded by the host designated agent.
5.19 Audit
When Patrons Execute audits, as a Part of Executing quality assurance, they Ought to Think about: 5.19.1 Purpose
The purpose of a host's audit, that will Be independent of and different from regular monitoring or quality control purposes, is to assess trial behavior and compliance with the protocol, SOPs, GCP, and the applicable regulatory requirements.
5.19.2 Selection and Qualification of Auditors
(a) The sponsor must appoint individuals, that are independent of their clinical trials/systems, to run research. (b) The sponsor should make sure that the auditors are qualified by experience and training to conduct audits properly. An auditor's qualifications must be recorded.
5.19.3 Auditing Procedures
(a) The sponsor should ensure that the auditing of clinical trials/systems is conducted with respect to the sponsor's written procedures about which to audit, the way to study, the frequency of analysis, as well as the shape and content of reports. (b) The host's audit program and processes for a trial should be directed by the value of the trial to admissions to regulatory authorities, the amount of subjects from the trial, and the form and complexity of the trial, and the amount of threats to the trial issues, along with also any identified issue (s). (c) The findings and observations of the auditor(s) ought to be documented. (d) To maintain the freedom and importance of the audit function, the regulatory authority(ies) shouldn't routinely ask the audit accounts. Regulatory authority(ies) could find entry to an audit report on a case by case basis if signs of critical GCP non-compliance is present, or even in the course of legal proceeding. (e) If required by applicable law or regulation, the host must offer an audit certification.
5.20 Noncompliance
5.20.1 Noncompliance with all the protocol, SOPs, GCP, or relevant regulatory requirement(s) with an investigator/institution, or from member(s)) of their host's staff should result in prompt action from the host to secure compliance.
5.20.2 in the event the observation and/or auditing describes long-term or serious noncompliance on the part of an investigator/institution, then the host must terminate the employee's /institution's involvement at the trial. Once an investigator's/institution's participation is terminated due to noncompliance, the host must notify immediately the regulatory authority(ies).
5.21 Premature Termination or Suspension of a Trial
When a trial is prematurely terminated or suspended, then the host should immediately inform the investigators/institutions, along with the regulatory authority(ies) of their conclusion or suspension and the reason(s) for the termination or suspension. The IRB/IEC also needs to be advised promptly and given the rationale (s) for the termination or suspension from the host or from the investigator/institution, according to the applicable regulatory requirement(s).
5.22 Clinical Trial/Study Reports When the trial has been completed or terminated, the sponsor should make certain that the clinical trial accounts are prepared and supplied to the regulatory agency(ies) according to the applicable regulatory requirement(s). The host must also make sure that the clinical trial reports in advertising programs meet the criteria of this ICH Guideline for Structure and Content of Clinical Study Reports. (NOTE: The ICH Guideline for Structure and Content of Clinical Study Reports reveal that abbreviated study reports may be appropriate in certain instances.)
5.23 Multicentre Trials For multicentre trials, the sponsor must make sure that:
5.23.1 All researchers conduct this trial from strict compliance with the protocol agreed to by the host and, if necessary, from the regulatory authority(ies), also awarded approval/favorable remark by the IRB/IEC.
5.23.2 The CRFs are made to capture the essential information at all multicentre trial websites. For those researchers that are collecting further information, supplemental CRFs must also be supplied that are intended to capture the extra data.
5.23.3 The duties of coordinating investigator(s) along with another participating investigators are recorded before the beginning of the trial.
5.23.4 All researchers are given directions on after the protocol, to complying with a uniform set of criteria for the evaluation of clinical and laboratory findings, and on finishing the CRFs.
6. But site specific advice might be given on separate protocol page(s), or handled in another agreement, and a few of the info listed below can be included in other protocol referenced documents, including an Investigator's Brochure. Any modification (s) must also bear the amendment number(s) and date(s).
6.1.3 Name and name of the individual (s) authorized to sign the protocol and the protocol change (s) for your host.
6.1.4 Name, name, address, and phone number(s) of their host's medical practitioner (or dentist when appropriate) for the offense.
6.1.5 Name and name of the investigator(s)) who is/are responsible for conducting the trial, along with the address and phone number(s) of the trial site(s).
6.1.6 Name, name, address, and phone number(s)) of the qualified physician (if appropriate), who's accountable for many trial-site associated medical (or dental) decisions (if other than investigator).
6.1.7 Title (s) and address(es) of the clinical laboratory(ies) and other technical or medical section (s) and/or associations involved with the trial.
6.2 Background Information
6.2.1 Title and description of the investigational product(s).
6.2.2 A list of findings in nonclinical studies that potentially have clinical significance and from clinical trials which are linked to this trial.
6.2.3 Summary of the known and possible risks and advantages, if any, to human subjects.
6.2.5 An announcement that the trial will be run in accordance with the protocol, GCP and the applicable regulatory requirement(s).
6.2.6 Description of the population to be researched.
6.2.7 References to literature and information which are related to the trial, which provide background for your trial.
6.3 Trial Objectives and Purpose
A comprehensive outline of the goals and the objectives of the trial.
6.4 Trial Design
The scientific integrity of this trial and the trustworthiness of the information from the trial depend considerably on the trial layout. A description of the trial design, must contain:
6.4.1 A particular statement of the principal endpoints and the secondary endpoints, if any, to be measured throughout the trial.
6.4.2 An outline of this type/design of trial must be performed (e.g. double sided, placebo-controlled( parallel design) and a schematic diagram of trial design, processes and phases.
6.4.3 A description of the measures required to minimize/avoid prejudice, such as: (a) Randomization.
6.4.4 An outline of the trial treatment(s) and the dose and dose regimen of the investigational product(s).
6.4.5 The expected duration of subject participation, and a description of this order and duration of all trial periods, such as followup, if any.
6.4.6 A description of the"stopping rules" or"discontinuation criteria" for different topics, elements of trial and complete trial.
6.4.9 The identification of any data to be recorded directly on the CRFs (i.e. no previous written or electronic record of data), also also to be regarded as source data. (b) The type and timing of this information to be collected for withdrawn subjects. (d) The followup to subjects withdrawn from investigational product treatment/trial therapy.
6.6 Treatment of Topics
6.6.1 The remedy (s) has to be treated, including the name(s) of the item (s), the dose(s), the dosing schedule(s), the route/mode(s) of government, and the treatment period(s), for instance, follow-up interval (s) for subjects for each investigational product treatment/trial therapy group/arm of this trial.
6.6.2 Medicine (s)/treatment(s) permitted (including rescue medication) and not allowed before or throughout the trial.
6.7.2 Methods and timing for assessing, recording, and assessing of efficiency parameters. 6.8.2 The timing and methods for assessing, recording, and assessing safety parameters. 6.8.4 The kind and length of the followup of subjects after adverse events.
6.9 Statistics
6.9.1 A number of the statistical techniques to be employed, including timing of any planned interim analysis(ses).
6.9.2 the amount of subjects planned to be registered.
6.9.3 the degree of importance to be utilized. 6.9.4 Criteria for the conclusion of this trial.
6.9.6 Procedures for reporting any deviation(s) from the original statistical plan (any form (s) from the original statistical plan ought to be clarified and justified from protocol or in the last report( as appropriate).
6.9.7 The choice of topics must be included in the investigations (e.g. all distinct subjects, all dosed subjects, all eligible subjects, evaluable subjects).
6.10 Direct Access to Source Data/Documents The host must ensure it is given in the protocol or other written agreement that the investigator(s)/association (s) will allow trial-related tracking, audits,
IRB/IEC inspection, and regulatory review (s), providing immediate access to supply data/documents.
IRB/IEC inspection, and regulatory review (s), providing immediate access to supply data/documents. 6.13 Data Handling and Record Keeping
6.14 Financing and Insurance Coverage Financing and insurance if not addressed in a separate arrangement.
6.15 Publication Policy Publication policy, if not handled in another agreement. 6.16 Supplements (NOTE: As the protocol along with the clinical trial/study document are closely linked, additional relevant information is found at the ICH Guideline for Structure and Content of Clinical Study Reports.)
INVESTIGATOR'S BROCHURE
7.1 Introduction
The Investigator's Brochure (IB) is a set of the clinical and nonclinical data on the investigational product(s) which relate to the analysis of the merchandise (s) in human subjects. Its objective is to deliver the researchers and others involved with the trial using all the data to facilitate their comprehension of the rationale behind, and their compliance with, several important features of this protocol, like the dosage, dosage frequency/interval, techniques of management: and security monitoring processes. The IB also gives insight to help the clinical direction of their research subjects throughout the course of this clinical trial. The info ought to be displayed in a concise, simple, objective, balanced, and also non-promotional type that allows an individual clinician, or possible investigator, to comprehend it and create his unbiased risk-benefit evaluation of the appropriateness of the planned trial. Because of this a medically qualified individual should normally take part in the screening of an IB, however, the contents of the IB must be accepted by the areas that created the described information. This guideline delineates the minimal information which needs to be contained within an IB and gives tips for its design. It's anticipated that the kind and degree of information available will change with the period of growth of the investigational item. If the investigational product is promoted and its pharmacology is broadly known by medical professionals, a comprehensive IB might not be vital. Where permitted by law enforcement, a fundamental product information booklet, package leaflet, or data could possibly be an proper choice, given that it comprises comprehensive, current, and comprehensive advice on all parts of the investigational product which may be of significance to this investigator. If a promoted product has been analyzed for a new usage (i.e., a brand new sign ), an IB unique to this new use ought to be ready. The IB must be evaluated at least annually and revised as required in accordance with a host's written procedures. More regular revision could be appropriate based on the phase of evolution and the creation of relevant new info. Nonetheless, in accordance with Good Clinical Nutrition, pertinent new information might be so significant that it needs to be conveyed to the researchers, and maybe into the Institutional Review Boards (IRBs)/Independent Ethics Committees (IECs) or regulatory governments before it's contained in a revised IB. Usually, the host is responsible for ensuring an up- to-date IB is made accessible for the investigator(s) and the researchers are responsible for supplying the up-to-date IB into the accountable IRBs/IECs. In the instance of a worker sponsored trial, then the sponsor-investigator must find out if a booklet is available in the industrial maker. If the investigational product is supplied by this sponsor-investigator, then they must offer the essential info to the trial staff. In scenarios when planning of a proper IB is impractical, the sponsor-investigator must supply, as a replacement, an enlarged background information element in the trial procedure which includes the minimal present data described within this principle.
7.2 General Considerations the IB should comprise:
7.2.1 Title Page
This ought to offer the host's name, the identification of every investigational product (i.e., study number, compound or accepted generic title, and transaction name(s) where legally permissible and desired by the host), along with also the launch date. It's also suggested an edition number, and a reference to this date and number of the variation that it supersedes, be supplied. A good instance is provided in Appendix 1.
7.2.2 Confidentiality Statement
The sponsor might desire to incorporate a statement instructing the investigator/recipients to take care of the IB as a private record for the only information and usage of this investigator's team along with the IRB/IEC.
7.3 Contents of the Investigator's Brochure
The IB should include these segments, each with literature references where appropriate: 7.3.1 Table of Contents An illustration of the Table of Contents is provided at Appendix 2
7.3.2 Summary
A short summary (preferably not exceeding two pages) ought to be granted, highlighting the substantial physical, chemicaland pharmaceutical, pharmacological, toxicological, pharmacokinetic, metabolic, and clinical data available that's pertinent to this point of clinical development of the investigational item.
7.3.3 Introduction
A short introductory statement ought to be provided that includes the compound name (and generic and trade name(s) when accepted ) of the investigational product(s), all active components, the investigational product (s) pharmacological category and its expected location in this category (e.g. benefits ), the justification for performing study using an investigational product(s), as well as the expected prophylactic, therapeutic, or diagnostic sign (s). At length, the introductory statement must offer the overall strategy to be followed in assessing the investigational item.
7.3.4 Physical, Chemical, and Pharmaceutical Properties and Formulation
A description ought to be given of this investigational product material (s) (such as the compound or structural formulation (e), plus a succinct summary ought to be due to the applicable physical, chemical, and pharmaceutical properties. To allow proper security measures to be obtained in the duration of this trial, an outline of the formula (s) to be utilized, such as excipients, ought to be supplied and justified when clinically applicable. Directions to the storage and management of this dose form(s) must also be granted. Any similarities with other substances should be noted.
ICH GCP E6 R2
On Mar 8, 2018, the FDA updated ICH E6(R1) with E6(R2) Good Clinical Practice: Integrated Addendum
to ICH E6(R1). Here are some noticeable changes and how they will impact the industry.
1. One of the key improvement is the new definition of a licensed copy of a situation report form (1.11). This improved definition states:"[a] newspaper or digital copy of the first document that's been confirmed (e.g., with a dated signature) or was generated via a validated procedure to make an specific copy using all the very exact features and data as the first." This improvement helps better explain how to ascertain the validity of trial-related documents duplicates, such as source files.
Additionally, the definition of tracking (1.38) has been broadened to incorporate the observation program, which can be described as "[an] outline of these methods, duties, and demands for tracking the trial." The updated definition doesn't call for the monitoring plan for a standalone file, but leaves an expectation that a proper plan is different. In addition, the observation report (1.39) definition has expanded to add "[results] of almost virtually any centralized monitoring also needs to be noted." Nonetheless many patrons now include concentrated monitoring as part of the general monitoring procedures because if this monitoring doesn't happen through a formal onsite observation trip, it might not be satisfactorily documented. The expanded definition will guarantee that patrons produce an account to demonstrate the concentrated monitoring that has been performed.
A number of improvements have been suggested to the Investigator department (part 4). for one, part 4.2.6 has been updated to say:"[in] the event the investigator/institution keeps the assistance of any party to do research jobs, they ought to ensure this celebration is qualified to execute these research jobs and should employ procedures to guarantee the integrity of their analysis jobs completed and any information created." These qualifications and oversight responsibilities weren't explicitly mentioned in the preceding edition, however, clinical trial sponsors anticipated researchers to implicitly stick to those guidelines. The upgraded statements today represent FDA's well- established advice on the pupil's supervisory responsibilities.
The definition of sudden adverse drug response (1.60) currently contains a brand new definition titled "identification of automatic systems" (1.60.1). But, there doesn't appear to be an evident connection between the definition of adverse medication reactions and this definition--"[a] practice of establishing and recording the specified prerequisites of a computerized system may be always fulfilled. An logical step will be to create this new PC validation definition 1.61 then renumber the past two definitions from the Glossary (vulnerable themes and well-being) into 1.62 and 1.63, respectively, and which could possibly be performed when the last record is published.
Part 5.18.6 (Tracking Report) contains a new section (e) that says "[tracking] results should be supplied to the host (such as proper management and personnel accountable for trial and website supervision) in a timely fashion for review and follow up as indicated. Outcomes of monitoring activities must be recorded in enough detail to permit confirmation of compliance with the observation program."
Section 5, Sponsors, comprises substantial adjustments and enhancements. The draft comprised a significant new segment 5.0 (Quality Management), where the notions of quality management, with a focus on risk management, are incorporated into the host's responsibilities. Though risk management procedures are well-known in the healthcare care sector, they have yet to be extensively applied to the preparation and execution of clinical trials. The upgrade will call for clinical trials patrons to start obtaining the essential instruction and tools to establish those principles. Two helpful overall resources include ICH Q9, Quality Risk Management, that will be a high level summary of risk management fundamentals, along with ISO 14971, Application of Risk Management to Medical Devices, a worldwide security standard related to all phases in the life span of a medical apparatus, for example its early growth. While these two records are tailored toward producing hazard management, they can provide useful information related to clinical trial preparation too. Table 1 lists the entire text of this suggested quality control department.
The draft creates no suggested modifications to the Department 3, Institutional Overview Board/Independent Ethics Committee.
In section 4.9, Records and Reports, a fresh introductory announcement (4.9.0) was added which says "[the] investigator must keep accurate and adequate source records and trial documents which have all applicable observations on each of the website's trial topics. Source data ought to be conducive, legible, contemporaneous, first, authentic, and complete. Changes to supply data ought to be traceable, shouldn't obscure the original entrance, and ought to be clarified if required (e.g., through an audit trail)."
Regular review of data that is submitted. Identification of lost information, conflicting data, information outliers, or sudden deficiency of variability and protocol deviations which could possibly be indicative of significant or systematic mistakes in data collection and reporting in a website or through sites, or might be indicative of possible data manipulation or data integrity issues. Utilization of statistical investigations to identify information trends like the consistency and range of information within and across websites. Evaluation of website features and performance metrics. Choice of websites and/or procedures are targeted onsite monitoring.
The previous modification increases section 8.1 (Introduction) the following improvements:"[the] host and investigator/institution need to keep a listing of the place(s) of the individual key documents.’ The storage method (no matter the media used) need to supply for record identification, research, and recovery. Based upon the actions being completed, individual trials will call for additional files not particularly mentioned in the vital document listing. The host or investigator/institution should incorporate these within this trial master document. The host should make sure that the investigator has command of continuous access to the CRF information reported to the host. The host shouldn't have management of these data. When a backup is utilized to replace a first record, the backup should meet the prerequisites for certified copies.
The draft includes several alterations that address fluctuations from the scale, sophistication, and expense of clinical trials because the former version was embraced. Since clinical research workers have access to innovative technologies and risk management procedures that might raise efficacy and concentrate on important clinical research actions, E6 has been amended to promote the implementation of advanced and more effective methods to clinical trial design, conduct, supervision, documenting, and reporting, while still ensuring human subject security and information integrity are preserved. Additionally, the upgrade includes changes to describe criteria on electronic documents and documents that are essential. In the end, the new record is intended to assist clinical research protect human areas, keep data integrity and quality, and correctly record trial benefits. This guide will emphasize the vital changes that impact research professionals. These alterations are anticipated to be assessed and approved inside ICH and then integrated into the E6 record from the end of 2016.
Revisions to the segment on tracking (5.18) reflect a stronger dependence on risk-based observation. The revisions include the components in the FDA's recent advice on risk-based observation. These alterations have been noted in part 5.18.3 (Extent and Nature of Monitoring) and comprise these improvements:"The host must create a systematic, guaranteed, risk-based method of tracking clinical trials. The flexibility at the scope and character of monitoring described in this section is meant to enable diverse approaches that enhance the efficacy and efficiency of observation. A combo of onsite and concentrated monitoring actions could be proper. The sponsor must file the rationale behind the selected observation approach (e.g., from the monitoring program )."
Part 5.20 (Noncompliance) comes with an augmentation which reflects regulatory ability expectations which patrons will try to recognize the root of non-compliance at a strong way and execute effective corrective and preventative strategies. The new segment (5.20.1) says:"[when] important noncompliance is found, the host must execute a root cause investigation and execute appropriate corrective and preventative actions. When required by law or regulation, then the host must notify the regulatory authority(ies) whenever the noncompliance is a severe violation of the trial procedure or GCP."
The draft also suggested a new segment 5.18.7 (Monitoring Plan) that says:"The host must develop a monitoring program that's tailored to the particular human subject security and information integrity risks of this trial. The program must describe the monitoring approach, the monitoring responsibilities of all of the parties involved, the a variety of tracking methods to be utilized, and the justification for their usage. The program should also highlight the observation of essential data and procedures. Particular care ought to be given to all these aspects which aren't regular clinical practice which need further training. The monitoring program should reference the related policies and processes."
Section 5.2, Contract Research Organization (CRO)also comprises two suggested changes that need sponsors to have a more active part in tackling their CROs. Section 5.2.1 was improved with the following announcement:"[the] host must ensure oversight of almost any trial-related responsibilities and works performed on its own behalf." Section 5.2.2 was improved with the following announcement:"[the] host must record approval of some subcontracting of all trial-related responsibilities and works with a CRO." This is very related to small and startup manufacturers which rely heavily upon CROs for handling all or most trial-related pursuits. The modifications state that patrons might not abdicate this duty and have to have a more active part in their supervision of the CROs.
Additionally, the ICH Upgrades underline the usage of centralized tracking as a vital approach to match and lower the frequency or extent of onsite observation.
Section 5.5.3 (b) is modified to describe expectations for normal operating procedures (SOPs) for digital data systems and handling. The proposed language says"[the] SOPs must pay for system installation, setup, and usage. The SOPs must explain system identification and performance testing, information collection and handling, program maintenance and system safety measures, shift management, information backup, recovery, contingency planning, and decommissioning. The obligations of the sponsor, employee, as well as other parties connected to the usage of those unmanned systems ought to be apparent, and the consumers must be supplied with instruction in the usage of their systems." The draft also comes with a brand new announcement 5.5.3 (h), which says that patrons are predicted to"[ensure] that the integrity of their information containing any information that explain the context, content, and arrangement of their information." This inclusion is very important whenever making modifications to the automatic systems, including software upgrades or migration of information.
Benefits of Getting Clinical Research Certification Online

Online Clinical Research Training Programs

Fig 1.1 Clinical Research Training Program
In 2024, amidst the constant hustle, maintaining a well-trained staff remains paramount for success and upholding integrity in clinical trials. However, amidst the bustling schedules, the importance of clinical research training often gets overlooked. Recognizing this challenge, online clinical research training programs emerge as a viable and cost-effective solution for teams spread across diverse locations, all deeply committed to advancing their research endeavors.
Despite the widespread availability of clinical research training institutes, many still struggle to access adequate training resources. To ensure your team stays productive and abreast of the latest developments, embracing online research training becomes imperative, even for seasoned researchers seeking to rejuvenate their expertise. Notably, online programs circumvent the expenses associated with travel, accommodation, and meals, rendering them more financially feasible than traditional in-person training sessions. Moreover, pre-recorded online classes offer scalability, obviating the need for hiring instructors for every session. For those looking to focus on specific roles within clinical research, programs like the Clinical Research Coordinator course and the Clinical Trials Assistant Training offer tailored training that fits into these needs.
In the face of pandemics or other logistical challenges, online training proves invaluable by streamlining scheduling complexities and bypassing the need for coordinating multiday workshops. The advantages of online training in 2024 are multifaceted:
Mobility Remote Learning
Easy Accessibility
Cost-Effectiveness
Flexibility
Constructive Criticism
Information Retention
Amidst the prevailing pandemic circumstances, numerous organizations now offer online clinical research certificates, tailored to deliver interactive training experiences through group activities, case study discussions, quizzes, and gamified learning approaches. For those interested in more specialized areas, consider exploring courses like the Pharmacovigilance Certification, CRA training, ICH-GCP Certification, or the Advanced Clinical Research Project Manager Certification. For physicians aiming at the forefront of clinical trial management, the Advanced Principal Investigator Physician Certification and Medical Monitor Certification offer routes to advanced expertise and leadership roles (Rachel Silver and Kessler, 2024).
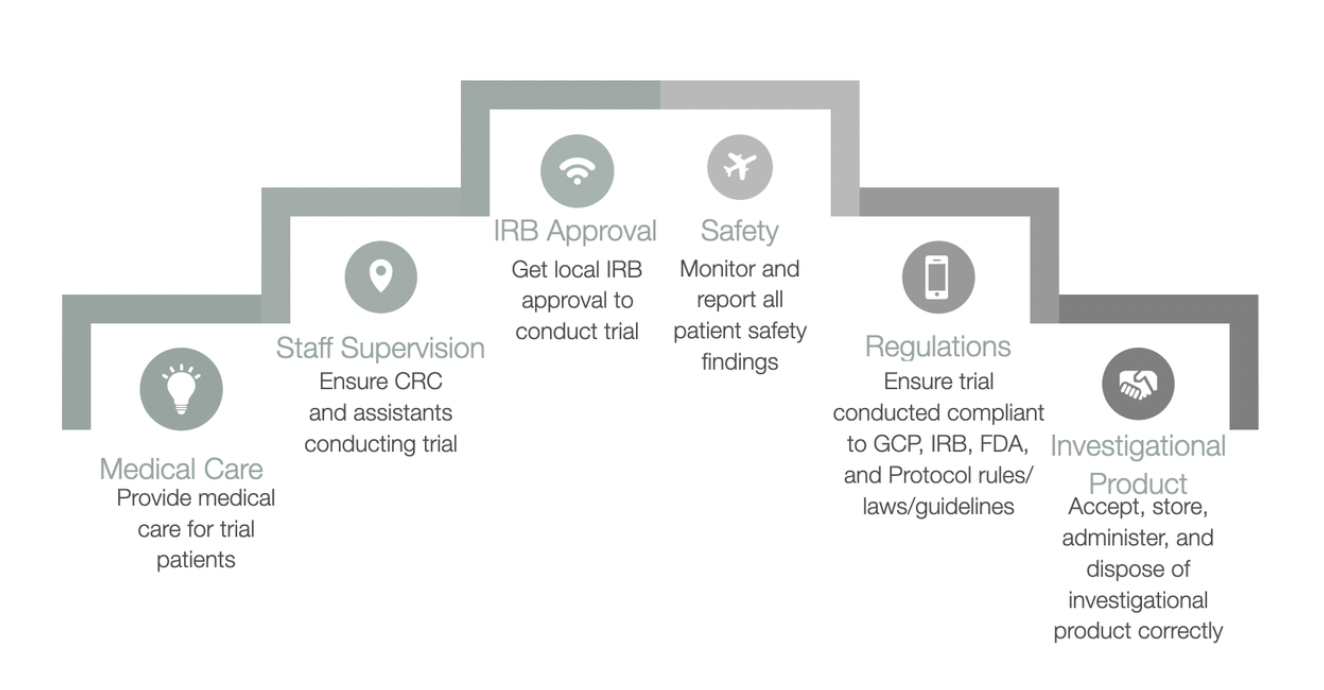
Fig 1.2: Training for Clinical Investigators
With the increased use of technology in education, online learning has now become a usual method. Today digital information is everywhere and is available for everyone. The use of online learning is increased since 2012, as evident by thriving online courses. Online learning, knows as internet-based learning, has no time and space limitations. Therefore making teaching and learning adorable by the internet-based delivery system. The effectiveness of online training is affected by many factors. Some factor creates a barrier for online training:
•Administrative issue
•Social interaction
• Technical skillet
• Cost and access to the internet
• Learner motivation
• Time and support
Below are many online certificate courses in clinical research online
Advanced Clinical Research Associate Certification (ACRAC)™
Advanced Clinical Research Coordinator Certification (ACRCC)™
Advanced Clinical Trial Assistant Certification (ACTAC)™
Group Orders & Organizational Training Subscriptions
Advanced Pharmacovigilance and Argus Safety Certification (APVASC)™
Advanced ICH GCP Certification (AGCPC)™
Advanced Physician Medical Monitor Certification (APMMC)™
Advanced Principal Investigator Physician Certification (APIPC)™

Fig 1.3 Clinical research training online, CCRPS
• CCRPS online courses: Training and continuing education from the society of clinical research associates that will promote quality research, protect the welfare of research, participate and improve global research.
• PI: Clinical research training for a spectrum of investigator and other involved in clinical research
Applied clinical research training programs offered by many organizations like CCRPS are for individuals seeking to enter clinical research to enhance their knowledge and help them required for employment. A clinical research training program helps to play an important role in our health care.
References:
https://www.imarcresearch.com/blog/why-now-is-the-time-to-invest-in-remote-clinical-research-training— WHY NOW IS THE TIME TO INVEST IN REMOTE CLINICAL RESEARCH TRAINING
https://www.med.unc.edu/crso/covid/other/remote-educational-opportunities-for-clinical-research-teams/ — Remote Educational Opportunities for Clinical Research Teams
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4477547/ — Applying Clinical Research Skills to Conduct Education Research: Important Recommendations for Success
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2829707/ — Defining Translational Research: Implications for Training
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6491176/ — E‐learning for health professionals
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6758693/ — Does online learning work better than offline learning in undergraduate medical education? A systematic review and meta-analysis
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5625951/ — Clinician–Investigator Training and the Need to Pilot New Approaches to Recruiting and Retaining This Workforce


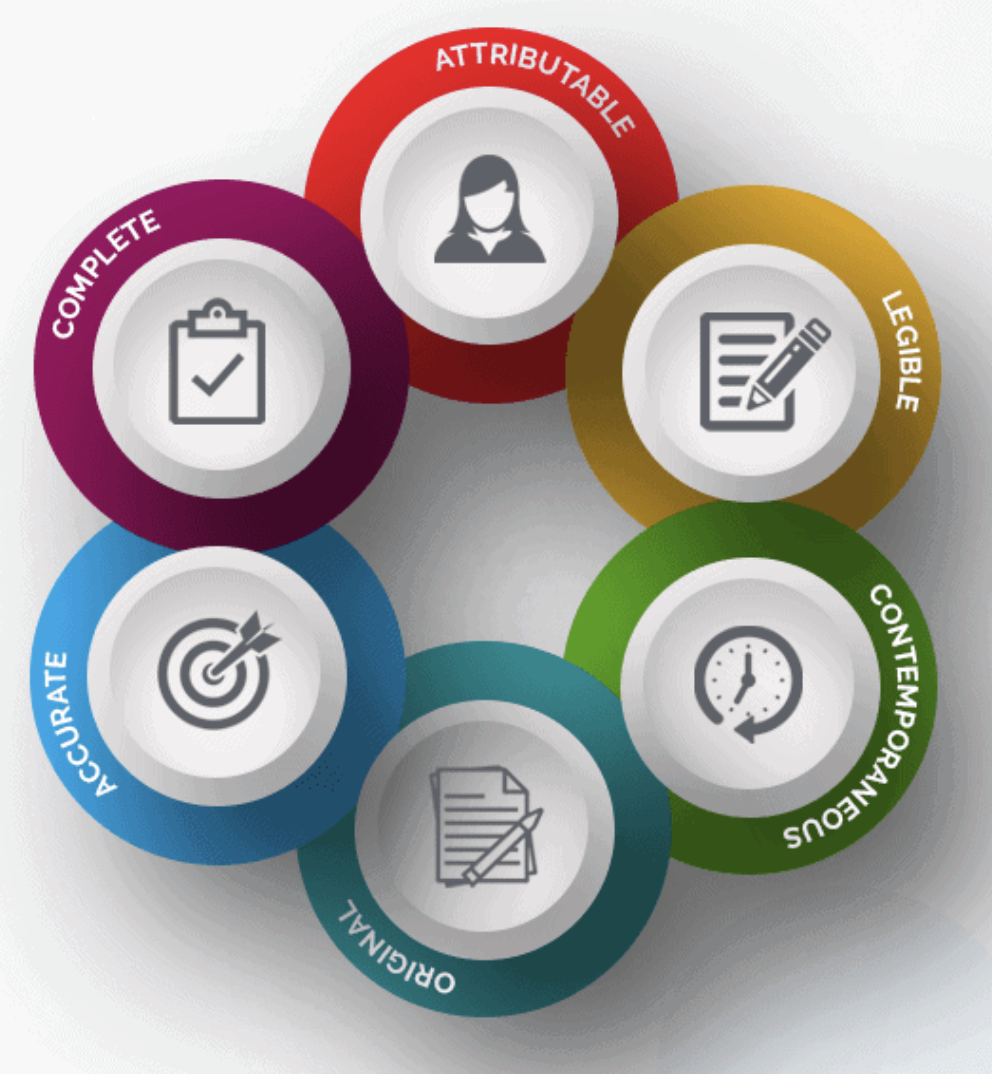






Clinical Research Associate Training and Placement
The role of the clinical research associate is very important in clinical trials to ensure that medical devices, new treatments and new drugs are approved for patients' use.
This field is taken as a certificate program course in many schools. You may also discover the availability of associate degree programs depending on the school. These programs can be completed in two years and can be offered through both the online and the hybrid formats. Hybrid formats combine both online and on-campus courses together.
If you opt for an online program, different platforms like emails and discussion boards are used to ensure and promote interaction between the students as well as with the lecturers. Online learning platforms are used to upload the syllabus, course materials, lectures and assignments. Some online programs include field work as part of their requirements, in order to gain first hand experience working with clinical trials and patients. Depending on the school, they may have a list of approved clinical research institutes and other facilities. Otherwise, you will have to find a facility for yourself and get the school's approval.
These certificate programs are generally designed for professionals that are already in the medical fields like medical assistants, nurses etc, and are interested in moving to the field of clinical research. They may therefore ask for a copy of your CV or resumé or they may ask for a letter from your employers to verify that you have the needed medical experience. Some programs may require just an undergraduate degree in a medical science or life science related field.
Clinical research associates are trained to assist clinical researchers and investigators in the coordination, administration and management of clinical trials. During this training, different courses will be taught revolving around subjects like safety procedures, subject recruitment, regulatory requirements, drug development, accountability, trial management, medical terminology etc.
The importance of the role of the clinical research associate means that companies that conduct clinical trials are usually very selective, the need to comply with strict regulations often inform their decision when making a choice of their clinical research associate. It is therefore very difficult to get a job as a clinical research associate without previous experience of clinical trials. Many companies require around at least two years experience in clinical monitoring as a clinical project assistant or clinical trial administrator before considering applicants for this important role.
In applying for the post of a clinical research associate, ensure that you read the job description and indicate or highlights the relevant experience on your curriculum vitae. Your cover letter should be specific to the company you're applying to. Do not use a one-for-all cover letter. Personalize your cover letter to each company and highlight the skills that fit the specific requirements of the role. Not all companies advertise their vacancies, so you can try to find out about other unadvertised vacancies.
Take courses from CCRPS and learn more on how to become a clinical research professional. Explore courses like Clinical Research Coordinator, Pharmacovigilance Certification, CRA, ICH-GCP, Advanced Clinical Research Project Manager Certification, and Advanced Principal Investigator Physician Certification.
Discover more from Clinical Research Training | Certified Clinical Research Professionals Course
Contact Us

What You Need to Know About Being a CTA (Clinical Trial/Research Assistant)
Get the right clinical research assistant job from CCRPS, Here you can get the all information about clinical research assistant job post. To know more visit us now!
The work of a CTA (clinical trial/research assistant) is one of extreme importance to the clinical research institute. CTAs work in a very busy department. To succeed, they needs to have a keen eye for details and an open mind to learn. They need to be able to ask the right question and find the right solutions.
DUTIES
The CTA helps in the collection and organization of data that is procured from studies. A typical day of a CTA is spent observing and communicating with the volunteers who are recruited to handle these kinds of studies. They are then responsible for analyzing and interpreting the statistics, which helps researchers realize their conclusions and results.
One of the critical tasks of a CTA is performing the different safety and quality checks within their unit. These checks are routinely carried out daily, weekly, or monthly. For example, standard equipment such as freezers and fridges are checked at least twice daily. This is important because they are used for storing samples and medications that needs to be kept in controlled temperature. Even a slight deviation from the controlled temperature can impact the validity of the result and the research.
Additionally, emergency equipment is checked on a daily basis. This ensures the safety of all the staffs and volunteers within the clinical research institute.
Another part of a CTA's job is to assist members of the team and deal with queries from members of the public. A CTA has to work in an administrative capacity and help with research paperwork. There are others who may have to perform some basic medical responsibilities like administering the medication for trials and even drawing blood.
In the midst of these many duties, it is very important that the CTA is good at multitasking. A good communication skill (both written and verbal) is also very important in this line of work.
EDUCATION
Most of the jobs in this area need a person to have a bachelor’s degree within the life sciences. However, there are some employers who require an advanced graduate degree, like a BMSc.
It is important for the clinical research assistants have a strong educational background in the administration of clinical trials. This means that you may have the kind of skills that help in organization. Having an interest in statistics and math can be an added advantage.
If you don’t have a degree in the science field and don’t want to go back to school, finding training and real experiences in clinical trials is going to help you hand a CTA position. At CCRPS, we offer effective courses and experiences you need to get your foot in the door. We have a CTA course and a free ICH GCP certification course, which teaches important skills that will help you succeed in clinical research. If you are looking to advance further, consider our Advanced Clinical Research Project Manager Certification or the Advanced Principal Investigator Physician Certification. For those interested in the crucial aspects of drug safety, our Pharmacovigilance Certification might be the right fit. Explore these to gain deeper insights and practical knowledge.
Take courses from CCRPS and learn more on how to become a clinical research professional.
Discover more from Clinical Research Training | Certified Clinical Research Professionals Course






Top 4 Clinical Research Books You Need To Read
With scientific discoveries and technological advancements comes knowledge. This knowledge defines the scope and practices of the different fields of professions that they apply to. Due to the continuous nature of medical research, the medical sciences are not also behind on scientific discoveries. More often than not, there is a new field knowledge that changes the way people do their job. The change could be something in something routine as checking the blood pressure of a patient or measuring the body temperature. A smart clinical research scientist knows that they have to ensure that they stay up to date with scientific discoveries to help patients as well as themselves.
If you have been offered a clinical research job or you are interested in one, one thing you should know for sure is that to be successful in this field, you need to know your stuff. It is true what they say, to remain relevant in any field is to remain knowledgeable.
To help your cause, we have made a list of 6 books that will help you succeed in the clinical research field.
Fundamentals of Clinical Trials, by Lawrence M. Friedman, Curt D. Furberg, David DeMets. This book looks into key issues like assessment, reporting of results, randomization, interpretation etc. This book introduces clinical trials to you beautifully, with real life examples used to explain key features of clinical trial.
Designing Clinical Research, by Dr. Stephen B Hulley, MD, MPH, Steven R Cummings, MD, Warren S Browner, MD. This book is an important book for doctors, pharmacists, nurses and all medical professionals involved in medical research. It explains useful methods for designing, funding and implementing clinical research.
Publishing and Presenting Clinical Research, Third Edition, by Warren S. Browner MD. This book contains the essentials of clinical trials and publication. This book will come in handy for those who wants to know more about organizing, delivering, and publishing the results of their research in the best way possible.
Practical Guide to Clinical Data Management, Third Edition, by Susanne Prokscha. One issue that always pops up in clinical research is how to manage the large volumes of ever increasing data. If you are already working in the field, you could no doubt relate. This is a task that can be rightfully described as “extremely time consuming”. This book gives powerful insights on current industry tactics on the use of Electronic Data Capture (EDC) for clinical research. This book will help you solve the age-long problem of managing voluminous clinical research data.
Honorable mentions
Basic Principles of Clinical Research and Methodology by S. K. Gupta
A Clinical Trial Manual from the Duke Clinical Research Institute. Kate Davis, Margaret Liu.
We have introduced many good reads for those who like to stay informed. Whether you are someone looking to get started in the field or someone who is looking for a refresher, these are the perfect places to start. If you need motivation to get reading, here are some clinical research positions to remind you of what is possible when you apply yourself.
If you prefer online classes with professional guidance, check out CCRPS’ certification courses. They offer one of the only major ACCRE accredited courses in the US. The courses are created by real, senior professionals who skip the nonsense and get right into what you need to succeed in the field.
To learn more about the role of a Clinical Research Coordinator, consider this Clinical Research Coordinator course.
Explore the realm of drug safety with our Pharmacovigilance Certification.
Dive into clinical research specifics with the CRA certification.
Understand the essential regulations with the ICH-GCP course.
Begin your journey in clinical trials with the Clinical Trials Assistant Training.
Enhance your skills with the Advanced Clinical Research Project Manager Certification.
For physicians aiming at leading research, consider the Advanced Principal Investigator Physician Certification.
Specialize in monitoring clinical trials with the Medical Monitor Certification.
Take courses from CCRPS and learn more on how to become a clinical research professional.
Discover more from Clinical Research Training | Certified Clinical Research Professionals Course







