

Adverse Event Reporting in Clinical Trials: A Step-by-Step Guide
When conducting clinical trials, ensuring the safety of participants is paramount. Adverse events (AEs) are any undesirable experiences associated with the use of a medical product or intervention in a clinical study. Monitoring and reporting these events accurately are not just ethical obligations but regulatory requirements that can significantly influence the success and credibility of a trial.
In this comprehensive guide, we’ll take a deep dive into adverse event reporting in clinical trials—understanding its importance, the step-by-step process for reporting, and key considerations to keep in mind. Whether you're a Clinical Research Coordinator (CRC), Clinical Research Associate (CRA), or part of a study team, having a solid grasp of this crucial aspect of clinical trials is essential.
What is an Adverse Event?
An adverse event (AE) is any unfavorable or unintended sign, symptom, or disease that occurs in a patient during a clinical trial. It may or may not be related to the treatment being studied. While adverse events can range from mild symptoms like a headache to severe conditions such as organ failure, it is essential to report them accurately for the safety of participants.
Key Points about AEs:
Severity Levels: AEs can range from mild to life-threatening, depending on the nature and impact on the participant.
Types of AEs: They can include any health complications, such as adverse drug reactions (ADRs), procedural complications, or pre-existing conditions worsened by the treatment.
Expected vs. Unexpected: Some adverse events are anticipated based on prior knowledge of the intervention, while others are unforeseen and may present new safety concerns.
Why is Adverse Event Reporting Important?
Adverse event reporting plays a pivotal role in ensuring the safety and well-being of participants in clinical trials. Beyond compliance with regulatory guidelines, it is a critical aspect of maintaining the integrity and credibility of a trial.
Key Reasons for AE Reporting:
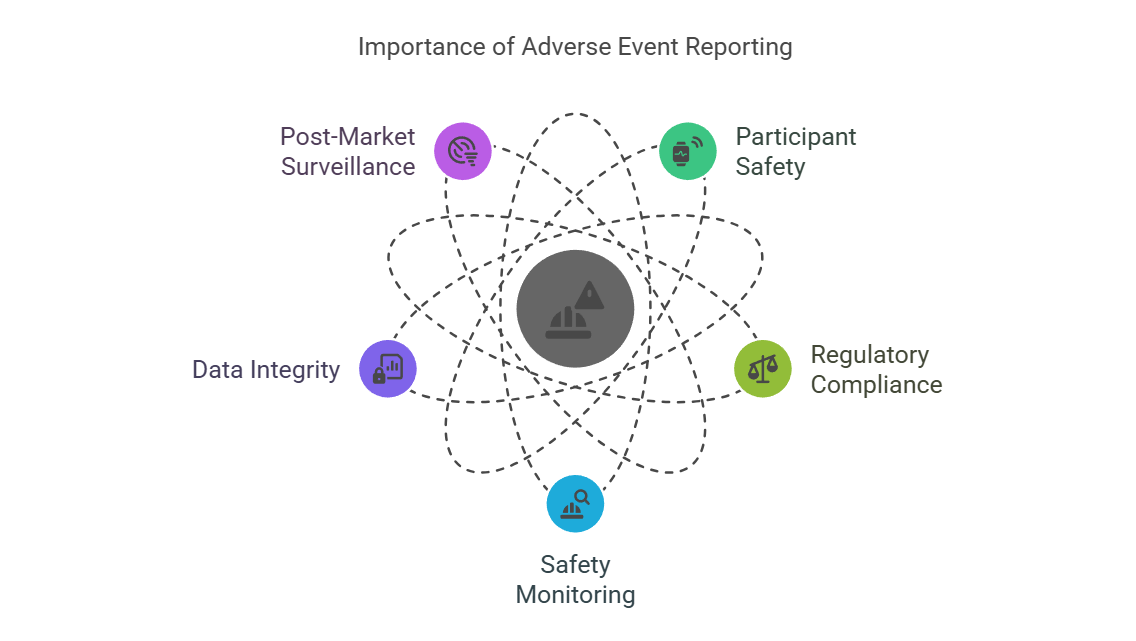
Participant Safety: Ensures that any serious issues affecting participants’ health are promptly addressed.
Regulatory Compliance: Failure to report AEs can lead to sanctions, halt of clinical trials, or revocation of product approval.
Safety Monitoring: Collecting AE data helps in identifying safety trends and enables the trial team to take corrective actions if needed.
Data Integrity: Accurate reporting of AEs maintains the integrity of the data, providing a clear picture of the treatment’s risks and benefits.
Post-Market Surveillance: AEs contribute to the broader post-marketing surveillance data, helping regulatory bodies like the FDA or EMA assess long-term safety.
Regulatory Requirements for Adverse Event Reporting
Several regulatory bodies have clear guidelines for adverse event reporting in clinical trials. In the United States, the FDA and the Code of Federal Regulations (CFR) play central roles in setting the standards for AE reporting, whereas the European Medicines Agency (EMA) oversees these activities in Europe.
Key Guidelines and Regulations:
FDA: The FDA mandates AE reporting for clinical trials under 21 CFR Part 312, which covers Investigational New Drug (IND) applications.
EMA: The EMA, through the Good Clinical Practice (GCP) guidelines, requires reporting of both serious and non-serious adverse events.
ICH GCP: The International Council for Harmonisation (ICH) provides a unified guideline that governs AE reporting standards globally, ensuring consistency across studies.
WHO: The World Health Organization (WHO) also has an active role in international safety reporting and the monitoring of global clinical trials.
Types of Reporting:

Serious Adverse Events (SAEs): These events require immediate attention and must be reported within 24 hours.
Non-Serious Adverse Events: While important, these do not require the same immediate attention but must still be documented regularly.
Step-by-Step Guide to Adverse Event Reporting
Adverse event reporting involves a meticulous process that needs to be performed in compliance with both protocol and regulatory guidelines. Below is a step-by-step guide that will help streamline the AE reporting process:
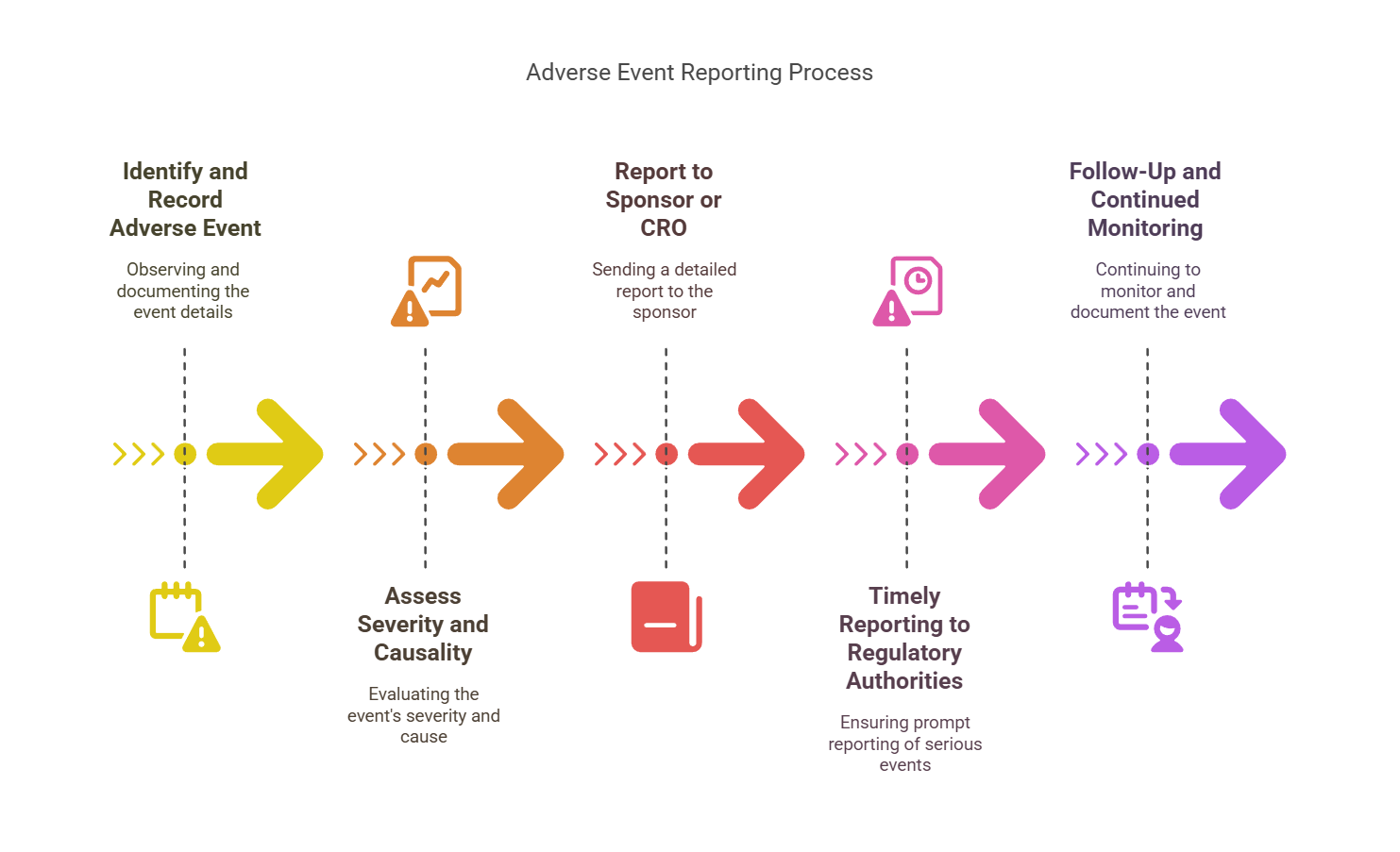
Step 1: Identify and Record the Adverse Event
Upon observing an AE, the first task is to accurately identify and document the event. Detailed records should be kept of the onset, duration, and severity of symptoms, as well as any treatments administered.
Step 2: Assess the Severity and Causality
Once the AE is identified, the next step is to assess its severity (mild, moderate, or severe) and its causality (whether the event is related to the investigational product or not). This assessment is typically done by the investigator in consultation with the study team.
Step 3: Report to the Sponsor or CRO
The event must then be reported to the study sponsor or Contract Research Organization (CRO). A standard form, such as the MedWatch form (for the FDA), is often used for AE reporting. The details of the report should include:
Patient information (without compromising confidentiality)
Date of onset and resolution
Description of the adverse event
Relationship to study drug
Outcome of the event
Step 4: Timely Reporting to Regulatory Authorities
Certain adverse events, particularly serious adverse events (SAEs), must be reported to regulatory authorities within specific timelines:
SAEs: 24 hours for serious, life-threatening AEs
Non-Serious AEs: Can be reported in periodic safety updates
Step 5: Follow-Up and Continued Monitoring
After the initial reporting, follow-up monitoring and continued documentation of the AE must occur. In some cases, further investigation may be needed to determine the cause or outcome of the AE. Additional reports may be filed as new information becomes available.
The Role of the Clinical Research Coordinator in Adverse Event Reporting
As a Clinical Research Coordinator (CRC), it’s your job to ensure accurate AE documentation and to serve as a liaison between participants, investigators, and the sponsor.
Key Responsibilities of CRCs:
Ensure Documentation: CRCs maintain accurate records of all adverse events in trial logs, patient charts, and case report forms.
Monitoring: Continuous monitoring of participants helps in early detection of adverse events.
Communication: CRCs play a key role in ensuring clear communication between the study team and regulatory authorities.
Training: Ensuring that all staff members are well-versed in AE reporting guidelines and procedures.
For medical students seeking research opportunities, explore research jobs for medical students and kickstart your career in clinical research.
Types of Adverse Events
Adverse events in clinical trials can be classified into various categories:

Expected vs. Unexpected AEs: Expected AEs are listed in the protocol, while unexpected AEs arise unexpectedly and may warrant further investigation.
Serious Adverse Events (SAEs): SAEs are life-threatening or result in severe disability or death.
Non-Serious Adverse Events (NSAEs): These may be mild or moderate in severity, such as a headache or nausea.
Adverse Drug Reactions (ADRs): A type of AE that is specifically caused by the drug or treatment under investigation.
Challenges in Adverse Event Reporting
Reporting AEs can present challenges, including:
Underreporting: Participants or staff might not report AEs due to a lack of awareness or fear of study withdrawal.
Misclassification: AEs may be wrongly classified, affecting the quality of data.
Inconsistent Timeliness: Delayed reporting can impact regulatory compliance and trial safety.
Best Practices for Effective Reporting
To ensure the most efficient and reliable reporting of AEs, consider these best practices:
Train Staff Regularly: Ongoing training on AE reporting can minimize errors and delays.
Maintain Clear Documentation: Detailed and accurate records ensure compliance and participant safety.
Encourage Transparency: Create an open environment where participants feel comfortable reporting symptoms.
How Adverse Event Reporting Impacts Trial Results
Accurate AE reporting can greatly influence the outcome of a clinical trial. Early detection of trends or unforeseen risks can prompt adjustments to the study protocol or even early termination of the trial for safety reasons. Proper AE reporting also helps in providing valuable information to regulatory bodies, ensuring the safety of future patients.
Pro Tip: If you're aiming for a fulfilling career in clinical research, our guide on top positions in clinical research can help you identify the best opportunities in the field.
Conclusion
Adverse event reporting is one of the most critical aspects of clinical trial safety and efficacy. As a Clinical Research Coordinator or member of a clinical trial team, understanding and implementing an effective adverse event reporting process is essential to ensure the safety of participants and compliance with regulatory requirements. By following the outlined steps and best practices, clinical trials can successfully identify potential risks, mitigate harm, and ultimately contribute to the development of safe and effective medical products.
At CCRPS, we offer specialized certifications and training for Clinical Research Coordinators and other professionals, equipping them with the knowledge to excel in their careers and contribute meaningfully to clinical research.
-
An adverse event in a clinical trial refers to any unwanted or harmful medical occurrence experienced by a participant during the trial, whether or not it's related to the investigational drug or treatment.
-
Adverse event reporting is critical for ensuring participant safety, maintaining the integrity of the trial, and complying with regulatory requirements. It helps to identify risks and enables timely action to protect participants.
-
Both clinical investigators and the trial sponsor are responsible for reporting adverse events. Investigators monitor participants and report adverse events to the sponsor, who ensures timely reporting to regulatory authorities.
-
An adverse event can be any unfavorable outcome, while a serious adverse event (SAE) refers to events that result in death, life-threatening situations, hospitalization, disability, or permanent damage.
-
Adverse events should be reported using specific forms like the FDA's MedWatch or ICH E6 guidelines. These reports should include the event details, its severity, the participant’s condition, and the actions taken.
-
Regulatory bodies such as the FDA or EMA require that serious adverse events (SAEs) be reported within a set timeframe, typically 7 to 15 days, depending on the severity and trial phase.
-
Failure to report adverse events in a timely manner may result in regulatory sanctions, trial delays, or the suspension of the clinical trial. It can also compromise the safety of participants.
-
Yes, adverse event reporting can prompt changes in the trial, such as dose adjustments, safety monitoring protocols, or even the termination of the trial if risks outweigh benefits.