
Clinical Trial Protocol Development: PI’s Comprehensive Guide
Clinical trial success doesn’t start at site initiation—it starts at the protocol. The protocol is the backbone of every clinical study, shaping not only patient safety and scientific validity but also regulatory approval and operational feasibility. Whether you're developing your first trial or refining your tenth, getting protocol development right is non-negotiable. Principal Investigators (PIs), especially, must drive precision across every aspect—from endpoint definition to ethical compliance—ensuring the document can hold up to both scientific scrutiny and real-world execution.
This guide offers an unfiltered, in-depth breakdown of clinical trial protocol development. You’ll learn exactly what needs to go into the protocol, how to align it with global regulations like ICH-GCP and FDA guidelines, and how to navigate operational challenges before they become failures. Whether you're a new PI or a seasoned researcher aiming to elevate your protocol design strategy, this walkthrough will arm you with the expertise needed to create protocols that are both audit-ready and execution-strong.

Understanding Protocol Design Foundations
A protocol is not just a study plan—it’s a binding document that defines every clinical activity, regulatory responsibility, and scientific outcome. Poor protocol design leads to trial delays, protocol amendments, patient dropouts, and even trial terminations. Every Principal Investigator must therefore begin with a mastery of the design foundations that drive clinical viability and compliance.
Role of the Principal Investigator
The Principal Investigator (PI) is not just a signatory—they are the architect. Their role begins with defining the scientific rationale and clinical objectives of the study. A good PI synthesizes therapeutic knowledge with operational foresight to craft a protocol that meets ethical, statistical, and practical expectations.
Beyond science, PIs must ensure that risk-benefit ratios are justified, study procedures are implementable at clinical sites, and that subject safety is embedded in the methodology. They’re also accountable for aligning the protocol with Good Clinical Practice (GCP) standards and ensuring downstream staff understand and implement the document faithfully.
This means anticipating what could go wrong—from recruitment hurdles to adverse event reporting—and building structure into the protocol that ensures the site team is prepared. A PI who doesn’t lead at the protocol stage is already setting the trial up for protocol deviations, delays, or ethical breaches.
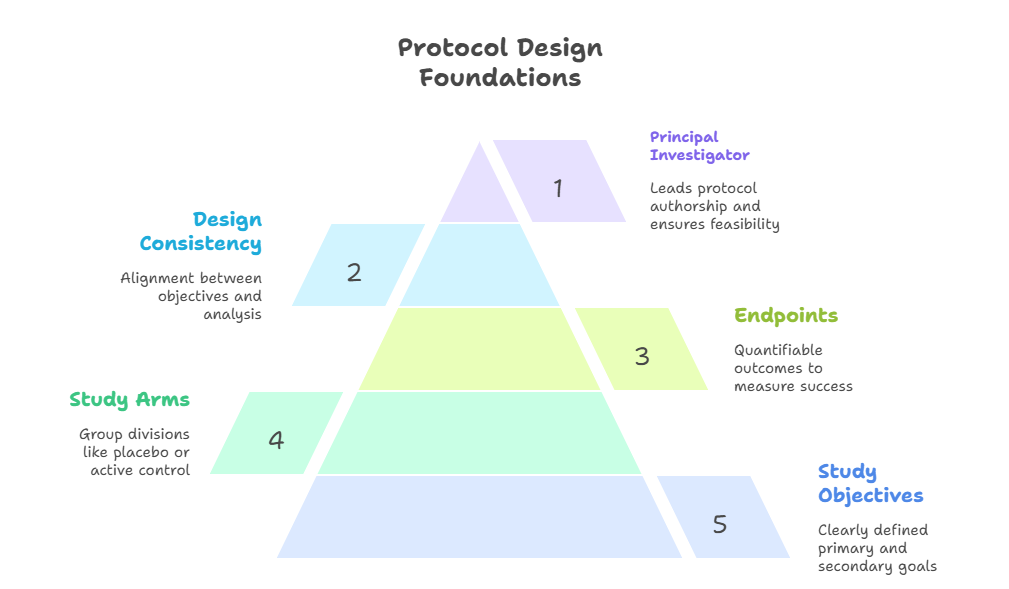
Key Protocol Elements (Study arms, objectives, endpoints)
A protocol is not evaluated based on length—it’s judged by precision, clarity, and regulatory alignment. The three pillars of any protocol are:
1. Study Arms
Define how subjects will be grouped—whether through placebo control, active comparator, or dose-ranging cohorts. Each arm must be scientifically justified and operationally distinct. Avoid overcomplication; too many arms create logistical burdens and statistical dilution.
2. Study Objectives
Objectives must be specific and hierarchical. Primary objectives drive the trial’s core purpose—often efficacy or safety—while secondary and exploratory objectives explore additional hypotheses. Regulatory bodies focus their review primarily on whether primary objectives are supported by validated endpoints.
3. Endpoints
Endpoints must be measurable, relevant, and time-bound. Primary endpoints determine whether the intervention works. These must be clearly defined (e.g., "reduction in systolic BP at 12 weeks") and paired with validated measurement tools. Avoid vague phrasing like “improvement” or “change,” as these are non-quantifiable and open to interpretation.
Each element above must be internally consistent with the inclusion/exclusion criteria, statistical analysis plan, and sample size calculations. A mismatch—such as defining an endpoint without proper power—will be a red flag for IRBs, sponsors, and regulatory authorities alike.
Clear, defensible protocols lower amendment rates, speed up site onboarding, and reduce patient risk. Getting these fundamentals right is where every successful trial begins.
Regulatory & Ethical Compliance Factors
Protocol approval isn’t just about scientific merit—it’s about demonstrating ethical and regulatory integrity across global and local standards. A protocol that fails to meet ethical oversight and international compliance doesn’t just stall your study—it invalidates your data. That’s why aligning your protocol with regulatory frameworks like ICH-GCP, FDA regulations, and institutional review standards is a mandatory, not optional, part of protocol development.
IRB and EC Requirements
Institutional Review Boards (IRBs) in the U.S. and Ethics Committees (ECs) globally serve as the ethical gatekeepers of clinical research. Their job is to ensure that human subject rights, safety, and well-being are upheld at all times. For a protocol to pass IRB or EC review, it must show robust risk mitigation strategies, clearly articulated informed consent processes, and scientifically justified study designs.
Protocols must describe every potential subject interaction—how, when, and why it occurs. Consent documents must mirror the protocol language, not paraphrase it. Additionally, all study procedures involving more-than-minimal risk must be accompanied by detailed safety monitoring plans. IRBs are increasingly rejecting protocols that provide vague language around AE management, placebo justification, or data confidentiality, especially in vulnerable populations.
Equally important is respecting local jurisdictional laws and cultural norms in multi-country trials. What passes ethical review in the U.S. may be flagged in Europe, Asia, or Africa due to differences in standards for compensation, consent age, or investigational treatment access post-study. PIs must either localize protocol language or build universal protections that exceed global minimums.
GCP, FDA, and ICH Guidelines Integration
Good Clinical Practice (GCP) is the global gold standard for conducting ethical and scientifically valid clinical trials. It governs everything from protocol design to documentation, investigator responsibilities, and monitoring requirements. Protocols that align with GCP are not just better designed—they are more likely to receive swift regulatory approval.
Under FDA regulations (21 CFR Parts 50 and 56), a protocol must clearly define risk minimization strategies, subject safeguards, investigational product accountability, and monitoring procedures. A protocol submitted to the FDA without a risk-benefit justification and endpoint rationale will likely be returned with a clinical hold request.
The International Council for Harmonisation (ICH) E6(R2) guidelines go further, requiring that protocols demonstrate quality-by-design principles. That includes integrating risk-based monitoring, critical data identification, and protocol deviation thresholds within the initial design. Rather than bolting these on later, successful PIs embed compliance into the structure from day one.
When protocols are pre-aligned with ICH-GCP and FDA frameworks, it accelerates IRB review, increases site adoption, and lowers the chance of costly protocol amendments post-launch. It’s the difference between a trial that crawls through red tape—and one that launches on time.

Operationalizing Protocol Execution
A protocol on paper is useless unless it translates into smooth execution in the field. Operationalizing a protocol means converting design into action—ensuring the study is not only compliant and scientifically sound but also logistically feasible and site-ready. This stage determines whether your protocol will run on time, recruit effectively, and generate data that regulatory authorities will trust. Without this, trials stall, protocols get amended, and costs balloon.
Feasibility Assessment (Site & Participant)
Site feasibility is not just about access to labs and equipment—it’s about capability, capacity, and alignment. A good protocol must anticipate whether sites can realistically follow procedures, enroll the right patients, and collect data as defined in the schedule of assessments.
Start with an internal feasibility grid:
Does the protocol require specialized equipment or skills that most sites don’t have?
Are the inclusion/exclusion criteria too narrow to enroll at a reasonable pace?
Are the visit windows, procedures, or sample handling expectations overly complex?
These misalignments cause site dropout and prolonged timelines. Even with trained investigators, a protocol that’s overburdened with dense procedures or rigid windows will struggle to execute without major deviations.
Participant feasibility is equally critical. PIs must evaluate whether the target population can and will comply with study demands—especially visit frequency, assessments, or lifestyle restrictions. For instance, a 12-visit, 24-week trial with multiple invasive procedures may work in a tertiary center but fail in community or rural recruitment zones.
Smart PIs pre-screen recruitment barriers, language needs, and transportation limitations during the protocol drafting stage, not after initiation. When feasibility isn’t assessed upfront, protocol amendments—and enrollment failures—are almost guaranteed.
SOP Alignment and Workflow Planning
Standard Operating Procedures (SOPs) form the backbone of how the protocol is executed across every site and vendor. However, most protocol writers neglect to cross-check if what’s being proposed can actually be implemented within existing SOPs. This results in miscommunication, compliance gaps, and data inconsistencies.
Workflow planning starts by mapping each protocol procedure into actionable tasks:
What happens at Visit 1, and who does it?
How is each biological sample processed and stored?
What are the escalation paths if a serious adverse event occurs?
Each of these steps must be mirrored in SOPs or require an SOP revision, and must be explained in operational documents like the Investigator Brochure, Manual of Procedures, and Site Initiation Training.
Equally important is timeline synchronization. For example, if your protocol demands interim analyses, is your EDC system programmed to lock and transfer that data on time? Is the data management vendor prepared to support those locks in real-time?
Failing to align protocol tasks with SOPs leads to friction between the CRO, sponsor, and sites—causing delays in monitoring, inconsistent data capture, and non-compliance findings in audits. A well-optimized protocol anticipates every downstream operational step, creating a blueprint that aligns scientific logic with execution logistics.
| Execution Factor | Details |
|---|---|
| Site Feasibility | Evaluates whether each site has the infrastructure, staff, and procedural readiness to conduct all trial-related activities. Assesses compatibility with equipment needs, visit schedules, and therapeutic area experience to reduce deviations and improve protocol adherence. |
| Participant Feasibility | Considers the subject population’s ability to comply with visit schedules, procedures, and follow-up. Addresses socioeconomic factors, travel burden, digital literacy, and language barriers to ensure realistic and sustainable enrollment. |
| SOP Alignment | Confirms that each protocol activity can be executed according to existing site and CRO SOPs. Identifies areas where new SOPs or protocol-specific guidance documents are required to ensure consistency across all trial locations. |
| Workflow Planning | Breaks down each visit and task into sequenced actions, assigning roles and responsibilities. Ensures clarity on timelines, data collection points, escalation paths, and source documentation requirements to avoid operational confusion. |
| Vendor Coordination | Aligns protocol timelines and expectations with third-party vendors managing data, labs, safety reporting, and logistics. Ensures proper communication pathways, role delineation, and contractual clarity for protocol-driven deliverables. |
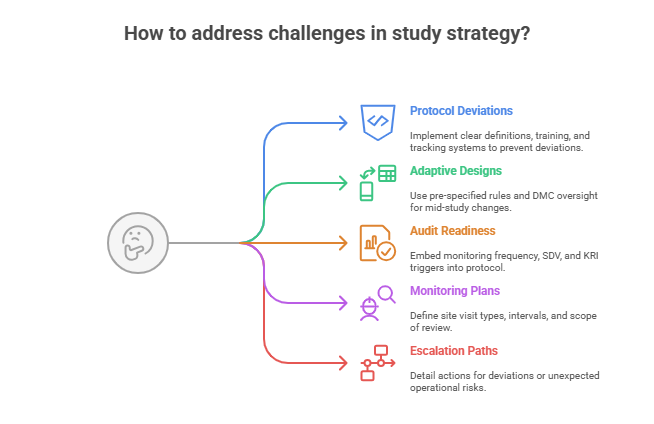
Real-World Challenges & Risk Mitigation
Even the most meticulously crafted protocols will collide with real-world obstacles. These include recruitment shortfalls, protocol deviations, noncompliance, and data integrity risks. The best PIs embed resilience into protocol design—anticipating challenges and coding in mitigation strategies that minimize impact and ensure trial continuity.
Adaptive Designs and Protocol Deviations
Modern trials rarely follow a straight line. Adaptive designs offer flexibility by allowing pre-planned modifications based on interim data—such as adjusting sample size, dropping ineffective arms, or reassigning randomization ratios. But adaptability must never compromise scientific rigor or regulatory integrity.
To succeed, adaptive protocols must include:
Statistically justified decision rules pre-approved by regulators
Clear thresholds for triggering adaptations
Tools for real-time data analysis and DMC (Data Monitoring Committee) oversight
Protocols that merely state “interim analysis will guide adjustments” without statistical scaffolding will be flagged by agencies and invite trial holds or post-hoc bias accusations.
Deviations, both major and minor, are another unavoidable reality. Common causes include:
Ambiguously written procedures
Visit scheduling conflicts
Unavailable lab tests or imaging equipment
Every protocol must include:
Deviation classification definitions (major/minor)
Predefined corrective action workflows
Real-time reporting and documentation structures
Proactively outlining these within the protocol reduces site confusion, streamlines monitoring, and prevents mislabeling of legitimate adaptations as noncompliance.
Audit-Readiness and Monitoring Plans
Protocol development must assume one thing: you will be audited—by sponsors, IRBs, CROs, or regulatory authorities. Building audit-readiness into the protocol makes you inspection-proof from Day One.
This means:
Incorporating monitoring frequency, visit types (remote, on-site), and scope
Identifying critical data points (e.g., informed consent, SAE documentation, endpoint validation) that will undergo 100% SDV (Source Data Verification)
Listing protocol-specific audit triggers such as recruitment delays, high deviation rates, or unblinded access events
Modern protocols should also specify whether risk-based monitoring (RBM) will be used, and if so, which Key Risk Indicators (KRIs) and Central Monitoring Analytics will support it.
For example, a trial assessing CNS endpoints may prioritize SDV for psychiatric evaluations, while a cardiovascular trial may emphasize lab values and ECG consistency.
Embedding monitoring and audit logic inside the protocol does more than reassure regulators—it empowers sites and CROs to operate confidently, reducing friction, delays, and rework.
A protocol that cannot be implemented cleanly, audited transparently, or corrected swiftly under pressure is not just a risk—it’s a liability. Anticipating these realities is what transforms a functional protocol into a resilient one.
Advanced Protocol Optimization Tactics
Clinical trial protocols are evolving rapidly. Sponsors and regulators now expect efficiency, adaptability, and digital compatibility—not just regulatory compliance. Optimization goes beyond writing clean procedures—it’s about engineering the protocol to support scalability, reduce subject burden, and enable high-integrity data across complex trial ecosystems.
eSource Integration, Decentralized Trials
Digitalization is no longer optional. Protocols must now be designed to accommodate electronic source data (eSource) and decentralized trial (DCT) methodologies from the outset. These aren’t add-ons—they directly influence how procedures are written, who performs them, and where they take place.
To integrate eSource:
Define which data points will be collected electronically (e.g., vital signs via wearable, patient diaries via app)
Specify how the eSource will be validated, secured, and integrated into the EDC
Align timing windows and audit trail requirements with 21 CFR Part 11 and GDPR
For DCT-ready protocols:
Identify remote-eligible procedures (e.g., televisits, at-home drug administration)
Clarify logistics for direct-to-patient shipments, remote AE reporting, and virtual PI oversight
Include technology fail-safes and backup procedures in case of device issues
The shift toward decentralized methods requires more than tech—it requires redesigning workflows and consent processes to match distributed, multi-device study designs. Protocols that fail to address these components risk delays during site training, IRB pushback, or outright noncompliance.
Stakeholder Collaboration Strategies
The best protocols are not written in isolation. Collaboration with downstream stakeholders—CROs, investigators, data managers, and regulators—is essential to crafting a protocol that can be implemented globally and consistently.
PIs should:
Involve sites in reviewing feasibility before protocol lock
Include data managers in defining critical variables and case report form (CRF) structure
Consult regulatory experts to align procedures with country-specific guidance
Collaborative development leads to protocols that are both scientifically robust and operationally grounded. It also reduces the frequency of amendments—each of which costs thousands of dollars and causes delays in enrollment, data cleaning, and submission.
Sponsors must also anticipate how vendors will interpret protocol instructions. A poorly defined endpoint could lead to multiple interpretations across vendors, triggering data harmonization issues, inconsistent patient reporting, or even trial failure.
When stakeholders co-author the operational DNA of a trial, the protocol becomes a shared contract—not just a compliance document. This elevates site motivation, reduces ambiguity, and accelerates trial timelines.
| Optimization Area | Implementation Strategy |
|---|---|
| eSource Integration | Clearly define which data points will be captured electronically (e.g., wearable devices, ePROs). Ensure systems are validated, Part 11 compliant, and integrated with the EDC. Include audit trail protocols and specify timing for data collection, synchronization, and source verification. |
| Decentralized Trials (DCT) | Identify procedures that can be conducted remotely (e.g., tele-visits, remote labs). Address logistics such as direct-to-patient drug shipping, virtual PI oversight, and home health nursing. Include contingency plans for technology failures and regulatory variance across regions. |
| Stakeholder Collaboration | Involve CROs, site investigators, data managers, and regulatory experts during protocol drafting. Seek feasibility input early to reduce future amendments. Ensure protocol language is consistent with downstream SOPs, CRFs, and site capabilities. |
| Risk-Based Monitoring (RBM) | Define KRIs (Key Risk Indicators), centralized analytics, and triggers for on-site monitoring. Specify what constitutes critical data, how it will be monitored remotely, and how deviations or trends will be escalated. Align monitoring frequency with risk level. |
| Amendment Prevention | Design protocols that are clear, feasible, and cross-validated with all operational stakeholders. Anticipate future adaptation needs and build flexibility into study windows, visit schedules, and eligibility criteria to minimize the need for formal amendments post-launch. |
Master Protocol Development with CCRPS’s Principal Investigator Certification
Every detail outlined in this guide—study design, regulatory compliance, stakeholder alignment—is covered in depth in the CCRPS Principal Investigator Certification. This isn’t a surface-level course. It’s a deep, career-elevating training built specifically for professionals who want to lead clinical trials at the highest level of quality and compliance.
Through this certification, you’ll master:
Designing GCP-aligned protocols that stand up to FDA, EMA, and IRB review
Creating protocols that balance scientific rigor and operational feasibility
Embedding adaptive design, decentralized methods, and risk-based monitoring from the start
Whether you’re transitioning into the PI role or want to sharpen your protocol authorship for complex, multi-site trials, this program equips you with the tools to lead.
This isn’t about checking regulatory boxes. It’s about transforming how you design, execute, and own your trials—so your protocols drive real-world outcomes, audit-readiness, and regulatory approvals.
Frequently Asked Questions
-
A clinical trial protocol must clearly define the study objective, methodology, inclusion/exclusion criteria, study arms, endpoints, and statistical analysis plan. It also needs detailed sections on informed consent, monitoring, safety assessments, and data handling procedures. Regulatory expectations—such as ICH-GCP compliance and 21 CFR Part 11 compatibility—must be addressed throughout. The protocol must serve as a comprehensive blueprint for investigators, outlining the flow of each subject visit and how data will be collected and reported. Oversights or vague sections increase amendment risks, ethical concerns, and site delays. A strong protocol isn’t long—it’s clear, auditable, and actionable across all study sites.
-
The Principal Investigator (PI) plays a central role in translating scientific hypotheses into operationally sound protocols. PIs define the study’s clinical rationale, endpoints, and ethical safeguards, ensuring procedures are feasible within site infrastructure and patient populations. They also oversee risk-benefit assessments, protocol alignment with SOPs, and adherence to Good Clinical Practice (GCP) standards. A PI’s insight helps preempt real-world challenges like recruitment hurdles or data collection failures. Collaborating early with sponsors, statisticians, and data managers ensures the protocol reflects both scientific integrity and executional viability. PIs aren’t just signatories—they’re protocol architects guiding trial success from day one.
-
A protocol that fails to meet regulatory expectations—like FDA, EMA, or ICH-GCP guidelines—may face serious setbacks, including clinical hold, delayed IRB approval, or trial termination. Regulators scrutinize protocol clarity, endpoint validity, patient safety plans, and monitoring structures. Missing justifications for risk-heavy procedures or poorly defined outcomes will raise red flags. Additionally, if a protocol lacks proper alignment with local laws or privacy regulations (like GDPR), it may be rejected in international settings. Regulatory misalignment can also lead to post-initiation amendments, which are expensive, time-consuming, and may undermine data credibility and subject trust.
-
Protocol deviations often stem from ambiguous procedures, unrealistic timelines, or misaligned site workflows. Minimizing them starts at the design stage. First, protocols should use precise, unambiguous language that mirrors SOPs and site capabilities. Inclusion/exclusion criteria must be medically justified yet practically implementable. Visit schedules should account for real-world barriers like transport or staff availability. The protocol should also include a predefined deviation classification system, mitigation strategies, and escalation protocols. Early training, real-time monitoring, and strong site communication help detect deviations early. A well-crafted protocol, combined with proactive oversight, keeps trials compliant and on track.
-
Adaptive trial designs allow pre-specified changes to be made based on interim results—without compromising trial integrity or statistical power. These might include sample size re-estimation, arm elimination, or dose adjustments. Adaptive elements must be clearly described in the protocol with decision rules, timing, and statistical thresholds. When properly executed, they reduce resource waste and accelerate patient benefit discovery. However, regulators require robust justification and safeguards to prevent bias. Adaptive protocols should also include Data Monitoring Committee oversight and predefined stopping rules. Used correctly, they bring flexibility and efficiency—but demand precise protocol integration and planning.
-
Integrating eSource changes how data is collected, monitored, and validated—making it critical to address in the protocol. Protocols should specify what data will be captured electronically (e.g., ePRO, wearables, remote vitals), who will validate it, and how it's integrated into the EDC system and source verification process. This also affects timing windows, audit trail language, and backup procedures for tech failures. Regulators require eSource systems to be compliant with 21 CFR Part 11, including user authentication, electronic signatures, and data traceability. Ignoring these details can lead to inspection findings, missed endpoints, or rejected datasets.
-
IRBs often reject protocols due to insufficient ethical safeguards, unclear risk-benefit justifications, or incomplete informed consent processes. Common red flags include ambiguous procedures, vague AE management plans, or inadequate protections for vulnerable populations. IRBs also expect that investigators understand and implement the protocol—not just sign off on it. Multisite trials with cookie-cutter protocols often fail if they don’t account for local cultural norms or logistical barriers. Failing to involve IRB specialists during protocol drafting or using generic language for subject protections are mistakes that significantly delay trial start or approval.
-
To lead trials as a Principal Investigator, you must demonstrate expertise in protocol development, subject safety, regulatory compliance, and site management. The CCRPS Principal Investigator Certification offers a robust, CPD-accredited program designed specifically for aspiring and current PIs. The certification covers everything from protocol authorship and ICH-GCP principles to risk-based monitoring and audit readiness. It also includes practical simulations and documentation templates used in real-world clinical research. Completing this certification gives you the credentials—and the operational insight—to manage complex studies and act as a regulatory-compliant, inspection-ready leader.
Final Thoughts
Protocol development is where clinical trials either succeed or stall. It’s not just about scientific documentation—it’s about designing implementable, ethical, and audit-ready frameworks that survive the real-world chaos of recruitment, data collection, and regulatory scrutiny. Principal Investigators who master protocol authorship aren't just contributors—they're the keystone of study success.
If you’re ready to lead trials that don’t just meet standards—but set them—investing in high-level training is essential. The CCRPS Principal Investigator Certification provides everything you need to build, lead, and optimize trials from the ground up. With global standards shifting toward digitalization, decentralization, and adaptive methodologies, the time to upgrade your protocol expertise is now.
You don’t get a second chance to launch a trial right. And it all begins with the protocol.