
Principal Investigator Responsibilities in Clinical Trials: Expert Guide
Principal Investigators (PIs) are the legal, ethical, and operational leaders of every clinical trial. Their signature validates the protocol. Their oversight ensures subject safety. Their judgment determines whether a study stays on track or collapses under regulatory scrutiny. From protocol approval to SAE reporting, the PI is accountable for GCP compliance at every level.
A missed deviation, poor delegation, or lack of documentation can trigger an audit or IRB rejection. That’s why today’s investigators must go beyond credentials — they need actionable training. The CCRPS Clinical Research Certification equips PIs with simulation-based learning, inspection-ready document workflows, and real-world compliance scenarios. It’s not about checking boxes — it’s about mastering oversight in environments where ethics and evidence intersect.

The Core Role of a Principal Investigator
The Principal Investigator (PI) is the central authority figure in every clinical trial — legally responsible, ethically accountable, and operationally in command. No study activity can begin or continue without the PI’s oversight. Their role spans protocol validation, subject safety, GCP adherence, and all regulatory-facing responsibilities.
Legal Accountability & Oversight
A PI is not a figurehead. They must personally review and approve the study protocol, sign off on IRB submissions, and take responsibility for every amendment, deviation, and final report. This includes form 1572 filings, regulatory correspondence, and site-level audit readiness. They act as the study’s official representative, capable of answering for any procedural failure. That legal burden means decisions cannot be deferred — the PI owns them, even when tasks are delegated downstream.
Ethical Responsibility to Subjects
The PI is also ethically responsible for participant protection. This includes ensuring informed consent clarity and ethical oversight, maintaining voluntary enrollment, and overseeing adverse event management. They must ensure that all site personnel are trained to uphold ethical standards — not just operational procedures. When trial complications arise, it's the PI’s job to respond decisively, prioritize safety, and notify oversight bodies.
In short, the PI isn’t just a manager — they are the ethical and operational conscience of the entire trial.
The Core Role of a Principal Investigator
Legal Accountability & Oversight: The PI holds ultimate responsibility for trial conduct, including protocol validation, regulatory filings (e.g., Form 1572), and IRB submissions. They personally sign off on every deviation, amendment, and final report. All site-level documentation must be audit-ready under their supervision.
Ethical Responsibility to Subjects: Beyond regulatory obligations, the PI safeguards participant welfare. They ensure consent is properly administered, adverse events are reported, and all site staff operate under GCP-aligned ethical standards. Their decisions prioritize subject safety, even amid operational pressures.
This role is not administrative — it’s regulatory, scientific, and ethical. The PI stands at the intersection of compliance and care, making them the most critical force behind every approved clinical trial.
Key Responsibilities Across the Trial Lifecycle
A Principal Investigator’s responsibilities aren’t confined to one stage — they evolve throughout the entire lifecycle of the trial. From feasibility to final closeout, the PI is expected to maintain control, insight, and compliance, even as tasks are distributed across the team. Oversight is not episodic — it’s continuous.
Pre-Trial Phase
Before the first subject is enrolled, the PI must conduct a thorough protocol review and documentation during planning. This involves understanding study objectives, inclusion/exclusion criteria, and statistical endpoints — then translating those into site workflows. The PI also oversees training sessions for site staff, verifies that equipment is calibrated and GCP-compliant, and confirms IRB approval status. These pre-trial actions ensure that no activity proceeds without regulatory clearance or proper delegation.
During the Trial
Once the study is active, the PI must verify protocol adherence at every turn. This includes real-time monitoring of recruitment, data entry, drug accountability, and adverse events. The PI is also responsible for reporting and managing adverse events, assessing their severity, and determining if they qualify as SAEs requiring expedited reports. If deviations occur — whether operational or safety-related — the PI must document, correct, and, if necessary, notify the IRB or sponsor.
Site visits, interim monitoring reviews, and regulatory inspections all fall under the PI’s watch during this phase. Transparency and responsiveness are key.
Post-Trial Duties
At closeout, the PI ensures all source documents are complete, CRFs are signed off, and final accountability logs are submitted. They also contribute to the writing of clinical study reports, publications, and sponsor-required summaries. Archiving responsibilities — including data storage and access logs — also remain under their jurisdiction. The trial doesn’t end until every file, report, and regulatory record is signed, stored, and audit-ready.
The lifecycle responsibility of the PI doesn’t taper — it intensifies as the study matures.
| Trial Phase | PI Responsibilities |
|---|---|
| Pre-Trial | Review and validate protocol, assess inclusion/exclusion criteria, oversee staff training, confirm IRB approval, calibrate equipment, and ensure regulatory clearance before trial initiation. |
| During Trial | Monitor recruitment, drug accountability, AE/SAE reporting, protocol compliance, and documentation. Handle IRB/sponsor notifications for deviations. Oversee site visits, inspections, and data integrity. |
| Post-Trial | Finalize CRFs, source documents, accountability logs, and study reports. Supervise data archiving, ensure audit-readiness, and support publication/reporting obligations. |
Delegation vs Direct Involvement – What PIs Must Never Outsource
Delegation is essential in clinical research — but it has limits. The PI is allowed to delegate certain trial-related tasks to qualified staff, but remains fully responsible for oversight, accuracy, and regulatory compliance. Knowing what can and cannot be delegated is critical to both GCP adherence and trial success.
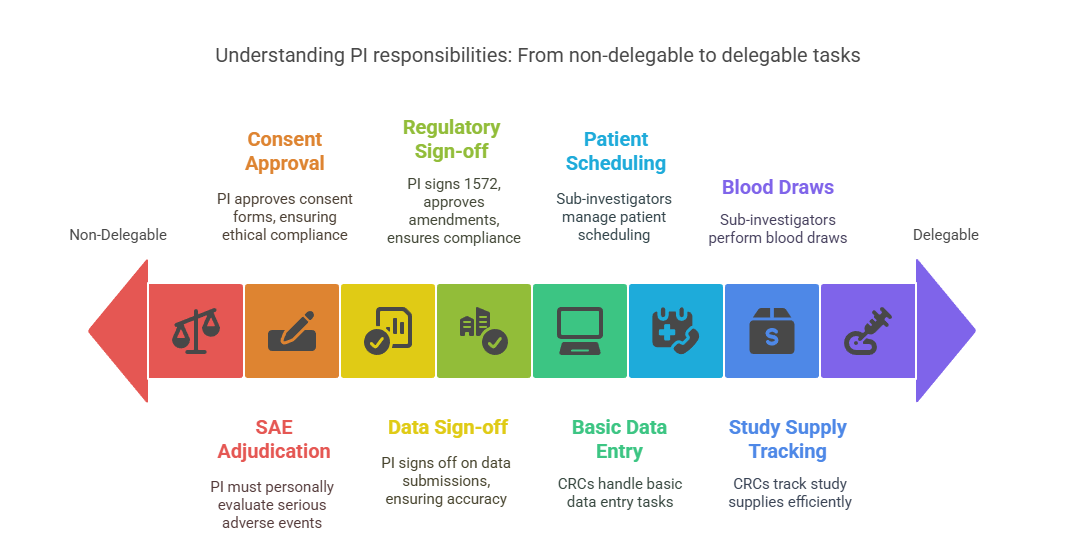
Tasks That Cannot Be Delegated
Certain decisions and responsibilities must always remain with the PI. These include final review and approval of consent forms, adjudication of serious adverse events (SAEs), and sign-off on all data submissions, especially CRFs. Regulatory bodies require that the PI personally evaluates subject eligibility, signs the 1572, and approves protocol amendments. These are legal responsibilities tied to the PI’s credentials and cannot be shifted — doing so could result in noncompliance findings during inspections.
Tasks That Can Be Delegated (with Oversight)
Tasks like blood draws, patient scheduling, basic data entry, or study supply tracking can be assigned to sub-investigators or CRCs. However, the PI must ensure proper training, oversight, and auditability of these activities. Delegation is not informal — it must be documented in a documented delegation and oversight log signed by both parties. The log must specify the scope, duration, and limits of each role. Even with delegation, the PI is still the one who answers to the sponsor, IRB, and regulators.
Effective delegation isn’t about offloading responsibility — it’s about managing it with structure, documentation, and constant review.
GCP Compliance and the PI’s Obligations
No PI can operate outside of GCP — it’s the regulatory standard that governs every action, from documentation to subject interaction. Being GCP-certified is not optional; it’s a core requirement for anyone leading a clinical trial. But understanding GCP isn’t enough — the PI must actively apply it across every stage of execution.
Understanding and Applying GCP
PIs must demonstrate current knowledge of ICH-GCP guidelines and FDA Part 11 compliance. This includes knowing how to respond to audits, conduct deviation assessments, and report adverse events within sponsor-defined timelines. Many noncompliance issues occur not because of bad intent, but because the PI delegated too much or failed to document oversight. To avoid these pitfalls, GCP principles must be applied to every site decision, whether large or small.
PI’s role in maintaining GCP standards also involves continual re-certification and training updates to stay current with evolving regulatory expectations.
Regulatory Documents the PI Is Accountable For
PIs must ensure that all essential documents are up-to-date, audit-ready, and properly signed. These include the delegation log, investigator CV, 1572 form, IRB approval letters, ICF templates, and source documentation. Oversight of source docs and deviation logs under PI review is not optional — it's a regulatory obligation. Errors or omissions in these documents have led to warning letters and trial suspensions.
A GCP-compliant PI is not just aware of documentation — they control it.
Which aspect of GCP compliance is most difficult for Principal Investigators to maintain?
Interfacing with IRBs, Sponsors, and CROs
PIs don’t operate in isolation — they are central to a web of communication that includes IRBs, sponsors, and CROs. Their ability to maintain transparent, timely, and well-documented correspondence directly affects site performance, audit outcomes, and study timelines.
Communicating with the IRB
Every protocol amendment, deviation, or unanticipated risk must be communicated to the IRB. The PI is responsible for ensuring that submissions are complete, justified, and delivered within required timelines. Whether it's a revised consent form or an SAE report, the PI must demonstrate IRB submissions and ethical oversight — failure to do so can result in halted enrollment or protocol suspension.
IRB communication also includes progress reports, continuing review documentation, and study closure forms. These must all be personally reviewed and certified by the PI prior to submission.
Working with Sponsors and CROs
Sponsors and CROs depend on the PI for status updates, data query resolution, and protocol adherence. During monitoring visits and audits, the PI must be available to clarify findings, explain site deviations, and confirm corrective actions. The PI’s responsiveness — especially in data clarification requests (DCRs) — plays a major role in whether a study remains on track or enters compliance escalation.
Professional, timely communication with sponsors and CROs is not just a courtesy — it is a regulatory expectation. The PI’s role is to keep all parties informed, compliant, and aligned on trial milestones.
Interfacing with IRBs, Sponsors, and CROs
Communicating with the IRB
- Submit protocol amendments and deviations with justification
- Ensure timely reporting of SAEs and consent updates
- Personally review and certify all IRB-bound documents
- Track progress reports and study closure filings
- Maintain ethical oversight via documented correspondence
Working with Sponsors and CROs
- Provide real-time site status updates and enrollment reports
- Respond to data queries and monitor findings proactively
- Support audit readiness through transparent site operations
- Clarify protocol deviations and confirm CAPA actions
- Ensure milestone alignment and regulatory responsiveness
PI Responsibilities by Phase: From I to IV
The PI’s role doesn’t remain static across different trial phases — it evolves with the risk, scope, and regulatory complexity of each stage. Understanding the specific demands of each phase ensures both ethical execution and GCP alignment.
Phase I – Risk Oversight & Escalation Doses
Phase I trials prioritize safety and dose escalation. PIs must monitor subjects intensively for early toxicity signals and pharmacokinetic responses. Every decision — from stopping rules to unblinding triggers — requires swift, evidence-based judgment. These trials demand the closest PI supervision, especially when using healthy volunteers or first-in-human compounds.
Read more: Phase I Clinical Trials – Explained: Objectives, Risks & Process
Phase II & III – Recruitment, Monitoring & Endpoint Evaluation
Phase II and III trials focus on efficacy, dose optimization, and statistical endpoints. The PI must ensure protocol-defined inclusion/exclusion criteria are strictly followed and subject eligibility is properly documented. Any deviation here can invalidate endpoints. During this phase, recruitment metrics, AE trends, and endpoint data require consistent PI involvement — especially as trial complexity increases and data volumes rise.
Explore:
Phase II Clinical Trials – Goals, Examples, and Real-Life Insights
Phase III Clinical Trials – Definitive Guide & Case Studies
Phase IV – Post-Marketing Surveillance
Phase IV requires the PI to track real-world safety signals, manage spontaneous reporting, and ensure proper patient education for long-term use. While enrollment may be broader and procedures lighter, the responsibility remains high — these studies often involve large populations and require vigilance for rare or long-term adverse events.
Dive deeper: Phase IV Clinical Trials – Post-Marketing Studies Clearly Defined
| Trial Phase | PI Responsibilities |
|---|---|
| Phase I Risk Oversight & Escalation Doses |
Prioritize safety and dose escalation protocols Monitor for toxicity and pharmacokinetic signals Make judgment calls on stopping rules and unblinding Oversee healthy volunteer and first-in-human participation Read more |
| Phase II & III Recruitment, Monitoring & Endpoint Evaluation |
Validate subject eligibility and protocol compliance Oversee recruitment, AE trend analysis, and endpoint monitoring Ensure strict application of inclusion/exclusion criteria Maintain data consistency as trial complexity increases Explore Phase II | Explore Phase III |
| Phase IV Post-Marketing Surveillance |
Monitor real-world safety and spontaneous AE reports Ensure accurate long-term patient education and use guidance Detect rare or long-term side effects across large populations Maintain pharmacovigilance and compliance reporting Dive deeper |
How CCRPS Prepares PIs for Full Role Mastery
Leading a clinical trial demands more than clinical knowledge — it requires regulatory precision, ethical leadership, and operational control. The CCRPS Clinical Research Certification equips Principal Investigators with the real-world tools needed to perform under FDA, EMA, and ICH-GCP expectations. This isn’t passive learning — it’s active skill-building.
Certification Tracks for New and Active PIs
Whether you’re a new investigator or stepping into global multicenter leadership, CCRPS offers structured certification tracks. Modules include protocol development, informed consent management, IRB oversight, and SAE reporting. PIs walk through every step of trial planning and execution — from site initiation to database lock. Each unit aligns with GCP, 21 CFR Part 11, and sponsor-level expectations.
The training emphasizes executional decision-making, teaching PIs how to respond to deviations, lead audits, and supervise cross-functional teams — while maintaining full regulatory control.
Applied Learning Through Role Simulations
CCRPS doesn’t rely on theoretical content alone. Investigators complete simulations that include oversight in clinical trial medical monitoring, deviation walkthroughs, audit prep scenarios, and mock sponsor visits. This applied learning ensures that trainees understand not just what to do, but how to do it — under pressure, with documentation, and in compliance.
PIs are taught how to handle real-world trial stress points: enrollment slowdowns, AE clusters, data discrepancies, and compliance flags. Each simulation strengthens decision-making confidence while reinforcing audit-readiness.
Practical Guides & GCP Alignment
Every module is built around current FDA guidance, GCP standards, and sponsor SOP expectations. Learners are provided with protocol templates, deviation logs, audit prep checklists, and consent review workflows — all of which can be immediately applied to real-world trials. More importantly, CCRPS teaches PIs how to lead ethically — aligning every operational decision with participant protection and protocol integrity.
By the end of the program, PIs walk away with more than a certificate — they gain mastery of the responsibilities that truly define successful investigators.

Final Thoughts
The Principal Investigator role is more than a title — it is a regulatory, ethical, and scientific obligation. Every trial, no matter how well-designed, depends on the PI’s vigilance to protect subjects, maintain GCP standards, and ensure data credibility. Their decisions influence everything: patient safety, protocol integrity, sponsor trust, and regulatory approval.
When a PI fails, the study risks collapse. When a PI leads with discipline and clarity, the trial thrives — even under scrutiny. That’s why certification isn’t optional; it’s foundational. The CCRPS Clinical Research Certification doesn’t just inform — it transforms PIs into operational leaders who can document, defend, and deliver at every phase of the trial. If you're responsible for trial oversight, mastering this role is your duty — and your edge.
Frequently Asked Questions
-
The PI is the legally and ethically accountable leader of the clinical trial site. Their responsibilities include protocol review, informed consent oversight, subject eligibility confirmation, SAE reporting, and regulatory document maintenance. They must ensure compliance with ICH-GCP, FDA guidelines, and sponsor SOPs. While tasks can be delegated, final oversight remains with the PI. They are the main point of contact during audits and IRB reviews and must be able to justify every decision made under their supervision. Their signature on the 1572 certifies site readiness, staff qualifications, and ethical standards compliance — making the PI’s role central to trial integrity and regulatory approval.
-
Yes, but with limits. A PI can delegate operational tasks like data entry, patient visits, or blood draws to qualified personnel. However, regulatory responsibilities such as consent form approval, SAE assessments, and final CRF sign-off cannot be delegated. Every delegation must be documented in a task log signed by both the PI and delegate. The PI must ensure staff are trained, qualified, and monitored. Even when tasks are handed off, the PI remains ultimately accountable for data accuracy, subject safety, and regulatory compliance. Improper or undocumented delegation can result in protocol violations and inspection findings.
-
Informed consent is one of the most ethically sensitive and legally binding components of any clinical trial. The PI must ensure that subjects fully understand the study’s risks, benefits, procedures, and their rights before participating. While others may assist in presenting the consent form, the PI is responsible for confirming comprehension, answering questions, and signing the documentation. Inadequate consent procedures can lead to IRB rejection, participant harm, or even trial suspension. The PI must also ensure that the most current, IRB-approved version of the form is used and stored in the site files.
-
The PI must maintain and oversee a range of critical regulatory documents, including the FDA Form 1572, IRB approval letters, delegation logs, ICF templates, CVs, training records, source documents, and protocol deviation logs. These must be up-to-date, audit-ready, and securely stored. Any changes — such as updated CVs or revised protocols — must be filed and documented promptly. The PI must ensure that documentation is not only complete but also traceable and reproducible, meeting both sponsor and regulatory requirements. Poor document control is a leading cause of noncompliance during site audits.
-
The CCRPS Clinical Research Certification provides PIs with practical, simulation-based training tailored to real-world investigator challenges. From protocol justification to IRB management and SAE reporting, the program ensures PIs are inspection-ready and compliance-driven. It includes mock audits, document walkthroughs, GCP refreshers, and decision-making drills under regulatory pressure. Unlike passive courses, CCRPS emphasizes operational execution and role accountability, preparing PIs to lead trials ethically, legally, and efficiently. Whether you’re new or experienced, the certification equips you with updated tools, templates, and SOP-aligned workflows essential for modern trial oversight.