
Top 20 Essential Terms for Regulatory Affairs Specialists in Clinical Trials
In the ever-evolving landscape of clinical trial compliance, mastery of essential regulatory terminology is not just beneficial—it’s critical for success. Regulatory Affairs (RA) specialists must navigate a complex web of global compliance frameworks, from ICH-GCP guidelines to the FDA’s 21 CFR regulations. Grasping key terms with precision enables faster submissions, minimizes errors, and ensures robust communication with health authorities. Without this fluency, even experienced professionals risk costly delays and regulatory pushbacks.
Understanding and applying these terms directly impacts submission accuracy, risk management, and drug approval timelines. A misstep in interpreting regulatory language can derail even the most promising studies. This guide dissects the top 20 must-know terms that define RA proficiency, combining real-world relevance with actionable insights. Whether you’re preparing a New Drug Application (NDA) or responding to an agency query, precise terminology is the backbone of effective regulatory strategy.

Top 20 Essential Regulatory Terms Table
Mastering the right terminology in regulatory affairs is non-negotiable. This table distills the 20 essential terms every RA specialist must know, each accompanied by definitions, practical usage, and real-world significance. Precision with these terms means faster approvals and fewer compliance errors.
| Term | Definition | Why It Matters | Usage Example |
|---|---|---|---|
| Investigational New Drug (IND) | Application to FDA to begin human trials of a new drug candidate. | Legally enables human testing, ensuring compliance and safety. | Filing an IND for a new oncology therapy. |
| New Drug Application (NDA) | Formal request for FDA approval to market a new drug. | Culmination of clinical development, unlocking market access. | Submitting an NDA post-Phase III trials for an arthritis drug. |
| Biologics License Application (BLA) | Request for FDA approval to market a biologic product. | Enables legal commercialization of biologics. | BLA filed for a novel monoclonal antibody. |
| ICH Guidelines | International standards harmonizing clinical trial and drug registration. | Streamlines global submissions, reducing redundancies. | Referencing ICH E6 (R2) in trial protocols. |
| Regulatory Compliance Plan | Structured approach to regulatory requirement adherence. | Minimizes risk, ensures proactive compliance. | Outlining audit prep and SOPs for a trial. |
| Clinical Trial Protocol | Document defining trial design, methodology, and analysis. | Blueprint for study conduct and submission success. | Drafting a protocol for a cardiovascular trial. |
| Clinical Study Report (CSR) | Comprehensive documentation of trial methods and results. | Key evidence for approval decisions. | Including CSR in an NDA submission. |
| Common Technical Document (CTD) | Standardized format for regulatory submissions. | Simplifies submissions to FDA, EMA, etc. | Compiling CTD modules for a new therapy. |
| Substantial Amendment | Significant protocol change requiring approval. | Ensures safety and data integrity. | Modifying inclusion criteria mid-study. |
| Serious Adverse Event (SAE) | Severe, unexpected reaction requiring immediate reporting. | Triggers rapid safety interventions. | Hospitalization from SAE during Phase II. |
| Orphan Drug Designation | Status for treatments targeting rare diseases. | Incentivizes rare disease drug development. | Orphan designation granted for pediatric therapy. |
| Clinical Data Interchange Standards Consortium (CDISC) | Data standards for trial data organization. | Facilitates transparency and regulatory review. | Formatting data using CDISC SDTM. |
| Data Monitoring Committee (DMC) | Independent group monitoring safety and efficacy. | Protects participants, maintains trial integrity. | DMC recommends trial halt due to safety issues. |
| 21 CFR Part 11 | FDA regulations on electronic records and signatures. | Ensures data integrity and legal compliance. | eRecords maintained per Part 11 standards. |
| Essential Documents | Critical documentation ensuring compliance. | Supports audits, inspections, and submissions. | Includes protocols, consent forms, and reports. |
| Quality Management System (QMS) | Framework for process quality control. | Reduces errors, bolsters regulatory confidence. | Implementing QMS for SOP management. |
| Good Clinical Practice (GCP) | Ethical and scientific quality standards for trials. | Protects participants, ensures valid data. | Mandatory GCP compliance for all submissions. |
| Clinical Investigator | Qualified physician overseeing trial site conduct. | Ensures protocol adherence and data integrity. | Principal investigator enrolls patients. |
| Post-Marketing Surveillance (PMS) | Monitoring drug safety post-approval. | Detects long-term or rare risks. | PMS through pharmacovigilance activities. |
| Marketing Authorization Application (MAA) | EU submission equivalent of NDA/BLA. | Secures EU market approval for therapies. | Filing MAA for an oncology drug with EMA. |
The Pillars of Regulatory Affairs Success
The Role of Regulatory Affairs Specialists
Regulatory Affairs specialists are the linchpin between clinical development and market access. Their expertise ensures that regulatory compliance standards are met while aligning submissions with the complex expectations of global health authorities. By meticulously preparing dossiers, managing interactions with agencies like the FDA and EMA, and staying ahead of evolving regulations, they mitigate risks and facilitate smoother approvals. These specialists are also responsible for monitoring post-marketing commitments and compliance audits, where even a minor lapse can result in severe penalties. Their strategic oversight spans from initial trial design to lifecycle management, ensuring that every step adheres to local and international compliance norms.
Beyond compliance, RA professionals play a pivotal role in optimizing time-to-market for new drugs and devices. They contribute to shaping study designs that anticipate regulatory requirements and maintain clear, accurate communication across interdisciplinary teams. With their sharp focus on detail, they translate scientific data into regulatory language that resonates with agencies. This enables seamless dossier preparation, reduces rejection risks, and enhances the credibility of submissions. In a globalized industry where regulatory landscapes shift rapidly, RA specialists provide stability, foresight, and expert guidance to navigate uncertainties and accelerate innovation.
Mastery of Regulatory Language
A deep understanding of regulatory terms isn’t just academic—it’s operationally critical. RA specialists must internalize precise definitions to navigate regulatory frameworks confidently. Misinterpreting a term like “substantial amendment” or “orphan drug designation” can cause delays, rejections, or compliance failures. Each regulatory term carries contextual nuances, dictating submission strategy and risk assessment. For example, knowing the difference between NDA and BLA streamlines application processes, while mastering clinical trial protocol language mitigates audit risks.
Moreover, mastery of terminology boosts interdisciplinary communication. Regulatory language bridges teams, from clinical operations to legal and medical affairs. Clarity in definitions ensures that all stakeholders are aligned, reducing miscommunications that could delay submissions or compromise study integrity. For RA specialists, this linguistic fluency is the difference between approval and rejection, between expedited pathways and drawn-out queries. It’s not optional—it’s a necessity for regulatory success.

Global Regulatory Frameworks That Shape Compliance
ICH Guidelines & Their Global Reach
The International Council for Harmonisation (ICH) guidelines, particularly ICH-GCP (E6 R2), serve as the global backbone for clinical trial standards. These guidelines harmonize expectations across regions, streamlining the development of clinical data packages that satisfy multiple regulatory bodies. By adhering to ICH standards, RA specialists avoid duplicative trials and conflicting regulatory responses, while ensuring data integrity and participant safety. This is crucial for global trials involving multiple sites and jurisdictions, where harmonized compliance reduces regulatory friction. Familiarity with ICH terms and structures also empowers RA teams to anticipate and respond effectively to evolving regulatory demands, accelerating time-to-market and enhancing approval prospects.
FDA & EMA Regulatory Landscapes
The FDA’s 21 CFR regulations and EMA’s Common Technical Document (CTD) format establish regional frameworks that, while aligned with ICH, introduce unique compliance nuances. The FDA’s focus on electronic records (21 CFR Part 11) and safety reporting mandates requires meticulous documentation and data integrity measures. Conversely, the EMA’s emphasis on modular submissions through CTD and centralized approval processes prioritizes standardization and cross-border transparency. RA specialists must master these region-specific requirements to tailor submissions effectively, ensuring compliance and minimizing the risk of delays or rejections. Mastery of these frameworks isn’t optional—it’s a strategic imperative for global regulatory success.

Common Pitfalls When Misunderstanding Key Terms
Submission Delays
A misstep in understanding key regulatory terms can trigger significant delays in submission approvals. For example, incorrectly applying a term like “substantial amendment” or “orphan drug designation” may require revisions or rework, prolonging the approval process. Regulatory agencies such as the FDA and EMA scrutinize every submission detail, and imprecise use of terminology can signal a lack of preparedness or comprehension. Each delay not only disrupts project timelines but also increases resource expenditure, creating operational bottlenecks. In today’s competitive landscape, where speed to market is a critical advantage, these setbacks can compromise a product’s commercial viability and market positioning.
Compliance Risks
Regulatory terminology is not just semantic—it’s a framework for ensuring compliance. Misapplying key terms like “Good Clinical Practice (GCP)” or “essential documents” can introduce gaps in data integrity, participant safety, and legal adherence. Non-compliance carries consequences, from warning letters to trial suspension or legal action. For instance, failure to follow 21 CFR Part 11 for electronic records can compromise data credibility, leading to penalties or rejection of submissions. Regulatory Affairs specialists must possess a granular understanding of these terms to proactively mitigate risks, maintain ethical standards, and safeguard both participants and organizational reputation.
Communication Breakdowns
Precision in regulatory language ensures clear, effective communication across cross-functional teams and with external regulators. Misunderstood or incorrectly used terms can result in misalignment between clinical, regulatory, legal, and operational teams. This misalignment often manifests in flawed protocols, ambiguous documentation, or ineffective responses to agency queries. Poor communication stemming from imprecise terminology not only slows down processes but also exposes companies to unnecessary risks. Regulatory Affairs specialists must bridge these gaps by mastering the language of compliance and embedding it into documentation, discussions, and submissions to maintain operational cohesion and regulatory confidence.
| Section | Key Points |
|---|---|
| Submission Delays |
|
| Compliance Risks |
|
| Communication Breakdowns |
|
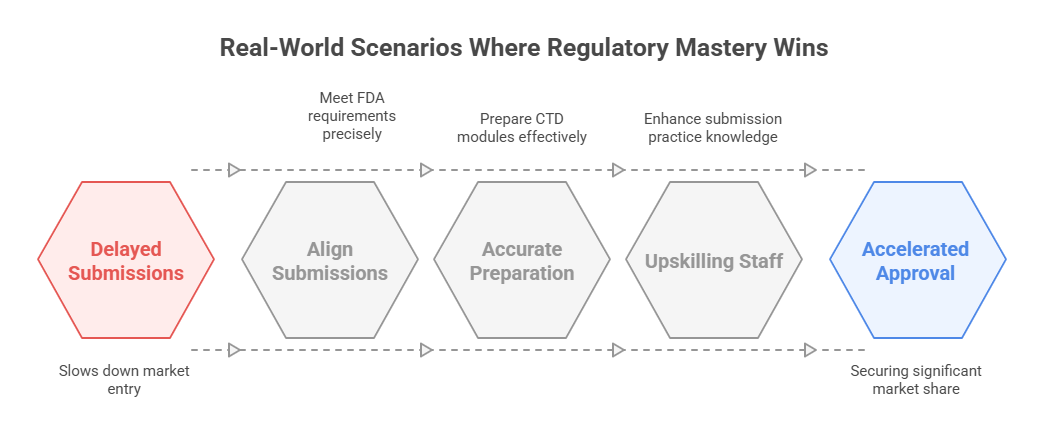
Real-World Scenarios Where Regulatory Mastery Wins
Case Study: Accelerated NDA Submission
A leading biotech firm, specializing in oncology therapeutics, faced aggressive timelines to submit a New Drug Application (NDA) for a promising new treatment. The Regulatory Affairs team’s deep understanding of key regulatory terms allowed them to align the submission with FDA’s stringent requirements, reducing back-and-forth queries and data clarifications. By meticulously preparing Common Technical Document (CTD) modules, ensuring precise terminology use, and integrating Clinical Study Reports (CSR) that met ICH-GCP standards, the company achieved a significantly accelerated review. The result: market approval was secured in a record timeframe, beating competitors to commercial launch and securing a critical market share advantage.
Lessons from Regulatory Hurdles
In contrast, a mid-sized pharmaceutical company faced setbacks during a biologics submission. Misunderstanding the distinction between Biologics License Application (BLA) and Marketing Authorization Application (MAA) requirements led to a misaligned submission to the European Medicines Agency (EMA). Additionally, insufficient grasp of CDISC standards for data formatting triggered multiple rounds of queries, delaying approval by several months. Recognizing these errors, the company restructured its Regulatory Affairs approach, investing in upskilling staff on key terms, enhancing data submission practices, and adopting a proactive communication strategy with regulators. The result was a streamlined resubmission, restoring trust and ultimately gaining approval—but only after costly delays and missed market opportunities.
How Our Clinical Research Associate (CRA) Certification Helps You Master These Terms
Achieving regulatory fluency isn’t just a professional advantage—it’s a career necessity. Our Clinical Research Associate (CRA) Certification, designed by CCRPS, delivers a structured, real-world-focused program that demystifies the top 20 essential regulatory terms critical for Regulatory Affairs Specialists. Unlike generic training, this certification goes beyond theory by embedding practical case studies, interactive modules, and scenario-based learning. It equips you with immediate, actionable strategies to navigate FDA 21 CFR, ICH-GCP, and EMA CTD requirements confidently.
Participants in the CRA certification master the nuances of submission frameworks, from New Drug Applications (NDA) to Biologics License Applications (BLA) and Marketing Authorization Applications (MAA). You’ll gain hands-on expertise in creating error-free Common Technical Document (CTD) modules, understanding CDISC standards, and aligning submission strategies to minimize delays. This certification also addresses high-stakes regulatory challenges, such as responding effectively to queries, mitigating compliance risks, and fostering cross-functional communication with clarity and precision.
The program integrates live webinars, mentorship, and a comprehensive curriculum covering ICH guidelines, regulatory frameworks, and submission strategies tailored for Clinical Research Associates and Regulatory Affairs professionals. Whether you’re new to regulatory affairs or seeking advanced mastery, this program provides the tools and confidence to excel in complex regulatory environments. You’ll be prepared not just to meet compliance standards, but to lead submission strategies, secure faster approvals, and drive innovation in your organization.
For detailed information on the CCRPS Clinical Research Associate (CRA) Certification, including course structure, features, and enrollment options, visit our dedicated course page at CCRPS CRA Certification. This certification doesn’t just teach you terminology—it transforms your understanding into a competitive advantage that accelerates your career and enhances your organization’s regulatory success.
Frequently Asked Questions
-
Mastering terms like Investigational New Drug (IND), New Drug Application (NDA), Biologics License Application (BLA), and Common Technical Document (CTD) is crucial. These define the framework for regulatory submissions and approvals. Terms like ICH Guidelines, 21 CFR Part 11, and Good Clinical Practice (GCP) establish global and regional compliance standards. Understanding definitions, context, and proper usage of these terms ensures accurate submissions, mitigates compliance risks, and streamlines interactions with regulatory authorities like the FDA and EMA. This fluency is not optional—it’s a core competency for Regulatory Affairs professionals managing clinical trial submissions and market authorizations.
-
Incorrect use of terms like “substantial amendment”, “orphan drug designation”, or “essential documents” can trigger rejections, additional queries, or even audit failures. Regulatory authorities, such as the FDA and EMA, scrutinize terminology closely. Misinterpretation signals a lack of preparedness, leading to delays in submission approvals. Each delay not only extends project timelines but also increases costs and jeopardizes market opportunities. To avoid this, Regulatory Affairs specialists must master precise terminology and embed it into documentation, communication, and compliance protocols, ensuring clarity, consistency, and faster approvals.
-
The CCRPS Clinical Research Associate (CRA) Certification offers comprehensive training in regulatory terminology, submission frameworks, and compliance strategies. It covers essential terms like IND, NDA, BLA, and CTD, along with detailed guidance on ICH-GCP standards and FDA/EMA regulations. CRA-certified professionals gain the skills to prepare accurate submissions, handle regulatory queries confidently, and ensure compliance throughout clinical trials. This certification is specifically designed to bridge knowledge gaps, enhance regulatory fluency, and fast-track career growth in Regulatory Affairs by equipping learners with both theoretical knowledge and real-world application.
-
ICH-GCP (International Council for Harmonisation – Good Clinical Practice) defines global ethical and scientific standards for clinical trials. It ensures participant safety, data integrity, and regulatory compliance. Adherence to ICH-GCP is mandatory for submissions to major regulatory bodies like the FDA and EMA. Regulatory Affairs specialists must interpret and apply ICH-GCP principles precisely in protocol design, documentation, and trial oversight. This minimizes compliance risks, improves approval timelines, and builds trust with regulators. Mastering ICH-GCP is not just about checking boxes—it’s about embedding a culture of compliance and quality throughout clinical development.
-
CTD modules standardize regulatory submissions across regions, reducing redundancies and enhancing transparency. They organize data into modules for quality, safety, and efficacy, facilitating simultaneous submissions to agencies like the FDA, EMA, and others. Mastery of CTD format ensures submissions are clear, consistent, and meet regulatory expectations. RA specialists proficient in CTD structure can anticipate and address agency queries proactively, reducing approval timelines. Whether preparing a NDA, BLA, or MAA, understanding CTD modules is vital for successful submissions in global markets.
-
In clinical trials, multiple stakeholders—from clinical teams to legal, medical, and regulatory functions—must collaborate seamlessly. Misunderstanding terms like “serious adverse event (SAE)” or “essential documents” can create confusion, misalign processes, or lead to non-compliance. Precise terminology acts as a common language that ensures everyone operates from the same playbook. Regulatory Affairs specialists bridge these gaps by embedding accurate terminology into protocols, reports, and communications. This not only enhances operational cohesion but also demonstrates to regulators a company’s commitment to quality and compliance, accelerating approvals and reducing operational risks.
Final Thoughts: The Regulatory Lexicon Advantage
Regulatory Affairs is a domain where precision isn’t optional—it’s the foundation of success. Mastering the top 20 regulatory terms isn’t just about vocabulary; it’s about wielding the tools that drive accurate submissions, reduce compliance risks, and accelerate time-to-market. Whether navigating complex frameworks like ICH-GCP, preparing CTD modules, or responding to FDA and EMA queries, fluency in regulatory language transforms challenges into strategic wins.
The CCRPS Clinical Research Associate (CRA) Certification empowers Regulatory Affairs professionals with the knowledge, skills, and confidence to thrive in high-stakes environments. By integrating practical case studies, expert-led webinars, and hands-on learning, this certification bridges the gap between theory and operational excellence. It’s your pathway to regulatory mastery—where every term you know, and every decision you make, contributes to seamless submissions and market success.
| Which aspect of mastering regulatory terms do you find most valuable? | |
|---|---|
| Streamlining submissions with accurate terminology | |
| Minimizing compliance risks through precision | |
| Enhancing cross-functional communication and clarity | |