
Clinical Research Project Planning: Essential PM Techniques
Clinical research project planning goes far beyond setting deadlines—it demands strict adherence to Good Clinical Practice, seamless team coordination, and vigilant safety oversight. From startup to closeout, every task must be aligned with regulatory standards and patient protection protocols.
The foundation of any successful plan begins with a well-structured clinical trial protocol. It defines the scope, objectives, and data strategy that drive the trial forward. Without protocol clarity, even the best timelines fall apart.
Equally critical is oversight at the site level. Effective investigator site management ensures documentation, consent processes, and compliance are maintained across regions—especially when scaling global trials.
This guide explores the essential project management techniques clinical teams need to navigate planning, mitigate risk, and deliver results with confidence.

Core Duties of a Medical Monitor in Clinical Trials
Medical monitors are the clinical safety gatekeepers of every trial phase. Their core responsibilities span across protocol adherence, safety surveillance, and ongoing collaboration with investigators. Without their vigilance, adverse events could go unnoticed and protocol deviations might compromise data validity.
Protocol Adherence and Oversight
Medical monitors ensure the trial is conducted exactly as outlined in the clinical trial protocol. They evaluate whether inclusion/exclusion criteria are followed, procedures occur on schedule, and dosing adjustments match protocol mandates. Monitors often conduct internal reviews to confirm alignment between planned procedures and actual execution.
Adverse Event and SAE Review
From headache reports to life-threatening SAEs, monitors must assess every incident’s relationship to the investigational product. They verify correct categorization, causality assessments, and timely reporting. For deeper understanding, refer to Adverse Events: Identification, Reporting, and Management.
Collaboration With Site Investigators
Clear communication with principal investigators (PIs) ensures real-time response to safety signals and data inconsistencies. Monitors coordinate with clinical staff to resolve data queries, support reporting obligations, and uphold investigator site management standards.
| Duty Area | Key Responsibilities |
|---|---|
| Protocol Adherence & Oversight | Ensure inclusion/exclusion criteria, scheduling, and dosing follow the approved protocol. Conduct internal reviews for procedural alignment. |
| Adverse Event & SAE Review | Evaluate all reported events for severity, relationship to the investigational product, and compliance with reporting timelines. |
| Collaboration With Site Investigators | Communicate with PIs to resolve safety issues and data discrepancies; ensure accurate query responses and investigator support. |
Regulatory Framework Medical Monitors Must Follow
Medical monitors play a key role in upholding the ethical and legal integrity of clinical trials. Their oversight spans not just clinical safety but also strict adherence to regulatory frameworks. These frameworks ensure that every aspect of a trial—patient enrollment, consent, treatment, and reporting—meets internationally recognized standards. Without this layer of governance, even the most scientifically sound protocol can face delays, disqualification, or liability exposure.
Good Clinical Practice & ICH Guidelines
One of the most critical regulatory anchors for medical monitors is Good Clinical Practice (GCP). GCP ensures that trials are conducted with full accountability to patient rights, data integrity, and ethical medical conduct. Medical monitors must actively assess whether investigators are applying GCP principles at every stage—from data collection to safety reviews.
Complementing this is the ICH Guidelines, a globally harmonized framework followed by most major regulatory bodies including the FDA, EMA, and PMDA. ICH E6 (R2), for instance, outlines the responsibilities of monitors and the importance of centralized oversight. Medical monitors must stay updated on evolving ICH revisions, as these directly impact how oversight is structured and what documentation is considered acceptable.
These frameworks do more than just set rules—they provide the legal and ethical blueprint that separates compliant trials from ones that risk suspension.
IRBs and Informed Consent Protocols
Before a study even begins, medical monitors are responsible for confirming that have thoroughly evaluated and approved the protocol. Without IRB approval, no patient recruitment or data collection is legally permissible.
Once trials are active, medical monitors ensure that informed consent is conducted properly at every site. This means checking that forms are version-controlled, approved by the IRB, and presented in a language and format that patients fully understand. They also confirm that re-consent procedures are in place for protocol amendments or new safety information. A failure to follow consent requirements can not only delay a trial—it can invalidate all data gathered from improperly consented patients.
In high-stakes therapeutic areas, such as oncology or rare diseases, medical monitors often serve as the final checkpoint between regulatory compliance and protocol deviation. Their vigilance ensures trials meet not just scientific goals, but also the ethical expectations of regulators, sponsors, and—most importantly—patients.
How prepared do you feel to handle emergency admin disruptions with task templates?
Oversight Across Clinical Trial Phases
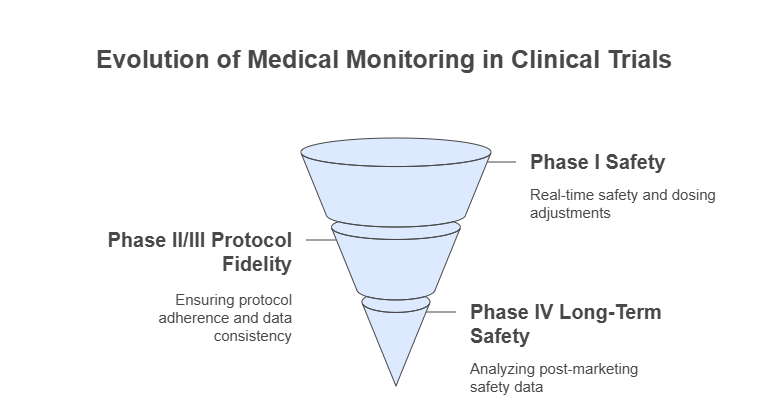
Medical monitors serve as the central safeguard for subject well-being and protocol fidelity across all trial stages. From first-in-human safety to post-marketing surveillance, their responsibilities shift—but never diminish. Knowing how oversight evolves phase by phase is critical for trial success.
Monitoring in Phase I–IV Trials
In Phase I clinical trials, medical monitors focus intensely on safety endpoints. They must review dosing tolerability, pharmacokinetic profiles, and any serious adverse events (SAEs) in real time. Early signals—like hepatic enzyme elevation or cardiac irregularities—require immediate interpretation and action. Monitors collaborate closely with principal investigators to adjust dosing schedules and inform go/no-go decisions.
During Phase II and III, monitors expand their scope. They ensure that clinical trial protocols are followed, patient eligibility is verified, and safety events are categorized accurately. Because these phases involve larger populations, oversight shifts from individual patient safety to broader data consistency and risk trend identification. Deviations, if left unchecked, can compromise regulatory approval.
In Phase IV trials, the focus pivots to long-term safety. Monitors analyze spontaneous adverse event reports, real-world evidence, and off-label use data. Their role becomes both reactive—managing safety issues—and proactive—identifying potential black-box warnings or market withdrawals. Even after drug approval, monitoring remains essential to protect patients and maintain public trust.
Evolving Responsibilities Across Trial Phases
The tasks of a medical monitor don’t just change—they mature. In early-phase trials, they operate more hands-on: guiding dose-limiting toxicities, interpreting pharmacodynamics, and steering safety board decisions. At this stage, the margin for error is razor-thin, and oversight is about real-time response.
In mid-to-late phases, responsibilities shift toward large-scale data analysis. Monitors compare AE frequency across populations, assess protocol compliance across dozens of sites, and support interim analysis decisions. Phase III trials, in particular, require intense coordination. Medical monitors help validate endpoints, monitor safety signal trends, and contribute key findings for FDA submission packages.
By post-marketing, their role is to synthesize signals from fragmented data sources—patient registries, spontaneous reports, EMRs—and advise on product labeling. If unexpected events arise (e.g., a rare cardiac risk in pediatrics), the monitor becomes the key decision-maker in label revision or product recall discussions. Their scope may now include regulatory engagement, representing safety data during advisory committee reviews.
Ultimately, the best monitors are those who shift smoothly from ground-level vigilance to system-level strategy—without losing sight of patient safety.
Quick Summary: How Medical Monitor Roles Evolve Over Time
- Early-Phase: Focused on dose-limiting toxicities, pharmacodynamics, and rapid-response safety actions.
- Mid-to-Late Phase: Shifts toward population-wide AE patterns, site compliance, and endpoint validation for submissions.
- Post-Marketing: Synthesizes signals from real-world data, supports label changes, and manages late-emerging risks.
Medical monitors must evolve from hands-on clinicians to strategic safety leaders—always with patient protection at the core.
Tools & Systems Used in Medical Monitoring
Modern medical monitors rely on digital ecosystems that provide real-time access to safety data, compliance status, and site activity. These tools aren’t just optional—they’re central to effective clinical oversight. From tracking adverse events to reviewing protocol deviations, these platforms allow monitors to manage complex studies with precision and speed.
CTMS and EDC Tools
Clinical Trial Management Systems (CTMS) and Electronic Data Capture (EDC) platforms form the backbone of clinical operations. CTMS platforms track patient enrollment, protocol adherence, and site performance in real time. For example, tools from the Top 20 CTMS systems offer dashboards tailored for monitors—flagging overdue safety reviews, enrollment lag, or unresolved queries at a glance.
Meanwhile, EDC systems enable secure, 21 CFR Part 11–compliant data entry and validation. Monitors use them to review lab results, adverse event reports, and concomitant medication data submitted by study coordinators. These tools allow rule-based data checks and real-time validation, minimizing errors and improving data quality across multisite trials.
When used in tandem, CTMS and EDC platforms allow monitors to cross-reference discrepancies between protocol schedules and recorded data—highlighting risk-prone sites, missing visits, or undocumented SAEs. This synergy supports early intervention and reduces the likelihood of downstream regulatory flags.
Safety Reporting & Signal Management Platforms
Beyond CTMS/EDC, medical monitors often rely on dedicated pharmacovigilance systems for detecting early risk patterns. These include software solutions that automate signal detection algorithms—tracking event frequency, severity, and trends across patient populations.
Many of these tools incorporate signal detection in pharmacovigilance directly into their core engine. When a particular event (like QT prolongation or hepatic dysfunction) surpasses the expected threshold, the system triggers an alert to the medical monitor for escalation. Some platforms also use machine learning to flag rare but serious patterns—such as event clustering by demographic or site.
Advanced safety systems also support global compliance by auto-generating MedWatch forms, EudraVigilance XMLs, or CIOMS reports—streamlining workflows and reducing human error. Monitors reviewing these signals must decide: Is this an isolated case, or the start of a safety trend? Their judgment here directly impacts sponsor decisions, IRB communication, and potentially, public safety.
Together, these platforms empower medical monitors to move from reactive reviewing to predictive safety management—ensuring higher trial integrity and participant protection.
| Tool Type | Purpose | Key Features |
|---|---|---|
| CTMS (Clinical Trial Management Systems) | Track trial operations, site activity, and enrollment | Real-time dashboards, overdue task flags, protocol schedule tracking |
| EDC (Electronic Data Capture) | Manage subject data securely and compliantly | 21 CFR Part 11 compliance, rule-based validation, AE data entry |
| Combined CTMS + EDC | Cross-reference data for protocol and safety alignment | Detect site-level issues, missing visits, and undocumented SAEs |
| Safety & Signal Management Systems | Monitor AE trends and trigger safety alerts | Signal detection, MedWatch/CIOMS form generation, machine learning risk flagging |
Skills & Qualifications for Medical Monitor Success
Becoming an effective medical monitor requires more than a medical degree. It demands a nuanced blend of clinical knowledge, regulatory expertise, and operational fluency across all trial phases. In high-stakes studies, even small misjudgments in adverse event handling or protocol oversight can delay approvals or endanger patients—making professional mastery non-negotiable.
Medical monitors are typically physicians (MD, DO, or equivalent) with a background in pharmacology, internal medicine, oncology, or other specialties aligned with the study. However, those transitioning into the role need structured training. The Medical Monitor Role Mastery course offers an end-to-end breakdown of responsibilities, from pre-study setup through post-marketing surveillance.
In addition to medical qualifications, monitors must be proficient in ICH-GCP, data review protocols, and risk-based monitoring principles. Familiarity with MedDRA coding, SUSAR assessment, and data query resolution is essential. For quick onboarding into clinical trial terminology, the Top 100 Clinical Research Terms resource is often recommended.
Communication is another critical skill. Monitors must translate technical safety findings into actionable insights for CRAs, data managers, and investigators. In sponsor meetings, they must defend medical rationales and guide safety decisions with clarity and confidence. Whether they're reviewing blinded data or presenting during a DSMB session, their authority stems from both expertise and trust.
To stay competitive, aspiring monitors should pursue continuous education, master modern tools, and develop cross-functional fluency. The best medical monitors aren’t just reviewers—they’re strategic allies in clinical success.

Final Thoughts
Medical monitors are the unsung anchors of clinical research—upholding patient safety, regulatory compliance, and scientific validity across every phase of a trial. Their oversight ensures that protocols aren’t just followed but critically evaluated, that adverse events are swiftly identified and escalated, and that every data point meets ethical and scientific standards.
In a world where regulatory expectations and trial complexity are rising, the role of a medical monitor has never been more essential. GCP adherence, cross-functional collaboration, and mastery of safety signal tools all define success in this high-stakes environment.
Yet it’s not just about vigilance—it’s about vision. Medical monitors who continuously upskill, adopt advanced technologies, and understand their influence across site, sponsor, and regulatory teams become indispensable leaders.
Whether you’re entering the role or refining your expertise, commit to excellence in clinical trial oversight. The impact of your judgment can shape approvals, protect lives, and move medicine forward.
Frequently Asked Questions
-
Most medical monitors are licensed physicians (MD, DO, or equivalent) with clinical experience, often in internal medicine, oncology, or infectious diseases. However, clinical research experience is just as crucial—especially familiarity with protocol design and GCP standards. Many professionals complete a Medical Monitor Role Mastery course to understand the ethical, operational, and regulatory duties of the role. Those transitioning from roles like sub-investigator, CRA, or safety physician typically succeed with additional training in AE evaluation and site engagement.
-
The Principal Investigator (PI) is responsible for conducting the trial at a specific site, while the Clinical Research Associate (CRA) monitors site performance and data accuracy. A medical monitor operates from the sponsor or CRO side, overseeing safety, protocol adherence, and medical questions across all sites. Unlike CRAs who focus on documentation, medical monitors assess clinical risk and provide input on adverse events, eligibility deviations, and trial amendments.
-
Medical monitors must master ICH E6(R2) Good Clinical Practice guidelines, including investigator obligations, safety reporting, and informed consent. They’re also expected to understand FDA regulations (21 CFR Parts 312, 50, and 56) and the operational role of IRBs and DSMBs. For foundational understanding, the What Is Good Clinical Practice (GCP)? and ICH Guidelines Simplified guides are highly recommended.
-
When an Adverse Event (AE) or Serious Adverse Event (SAE) occurs, site investigators report the data through EDC or email channels. The medical monitor evaluates the case, determines causality, and recommends follow-up or discontinuation. If life-threatening or unexpected, the event is escalated to the sponsor’s safety board and reported to regulatory authorities. Training in Signal Detection in Pharmacovigilance tools helps monitors distinguish isolated cases from broader safety trends.
-
Yes—medical monitors often lead safety sessions during investigator meetings, especially during site initiation visits (SIVs) or protocol amendment rollouts. They clarify medical nuances, answer eligibility questions, and provide SAE escalation pathways. These meetings are essential for aligning safety expectations across global sites. Participation helps reinforce protocol adherence and boosts site engagement, minimizing downstream deviations and audit risks.