
Clinical Research Associate: A Full Guide on Becoming A CRA in 2025

Whether you are new to this field or looking to advance your career, this article covers every aspect of becoming a successful CRA, including resources and practical advice for getting started.
What Does a Clinical Research Associate Do?
A Clinical Research Associate (CRA) plays a crucial role in ensuring that clinical research courses, clinical research certification, and clinical research certificate programs online adhere to ethical standards, regulatory requirements, and Good Clinical Practice (GCP) guidelines. But don’t stop with the basics—understanding the nuances of the role will give you a professional edge.
Core Responsibilities of a CRA

Monitor Clinical Trial Sites: CRAs ensure trials follow protocols by conducting site visits and remote monitoring. This is essential for professionals pursuing a clinical research associate course or looking for a clinical research associate certificate.
Ensure Data Accuracy: They validate and review trial data to maintain precision and completion, which is a key part of clinical research training.
Maintain Compliance: CRAs ensure trials align with global regulatory guidelines such as FDA, EMA, and ICH-GCP, which is covered in a clinical research certification program.
Protect Participant Safety: Oversee participant accommodations, clinical research requirements, and their overall well-being to meet certified clinical research associate standards.
Provide Professional Support: CRAs offer hands-on guidance and training to site staff, ensuring their operations comply with trial protocols. Enrolling in a cra training program can help individuals prepare for this responsibility.
Lesser-Known Tip: Developing a working knowledge of decentralized trials and using digital platforms like wearable monitoring devices can set you apart as an innovative candidate.
What is CRA Meaning in a Medical Context?
In a medical context, "CRA" stands for "Clinical Research Associate," which refers to a healthcare professional responsible for overseeing clinical trials, ensuring they are conducted properly by monitoring study sites, reviewing data, and ensuring compliance with regulations during the research process.
What Does a CRA Do? They uphold the structure that makes groundbreaking medical advances possible.
Best Clinical Research Associate Training Programs
Are you ready to jumpstart your career? The Best Clinical Research Associate Training Programs combine flexibility, practical knowledge, and real-world application.
Top Picks:
CCRPS CRA Certification
A fully customizable and self-paced course designed for both beginners and advanced learners.
IAOCR Certification Programs
Offers international focus, great for professionals interested in multi-national studies.
IQVIA CRA Training Program
Ideal for aspiring CRAs who want to specialize in tech-forward monitoring.
Mayo Clinic IATA Training
Focuses on niche skills such as biological sample transport, making it a great complement to traditional CRA learning.
Lesser-Known Tip: Supplement your training with hands-on modules or internships for tangible accomplishments on your resume.
CRA Certification Requirements
To become a Certified Clinical Research Associate (CCRA) or Certified Clinical Research Professional (CCRP), you'll need a bachelor's degree, relevant work experience, and industry-specific training. But what are the CRA certification requirements?
Here’s a breakdown:
Education Requirement: A Bachelor’s in life sciences is ideal (but not mandatory with prior experience or certifications).
Training Completion: Enroll in programs like CRA Training Program to fulfill competency gaps.
Monitor Hours: Certain certifications, like ACRAC Certification, may require logged hours of clinical trial monitoring.
Certifications like Clinical Research Associate Certification will elevate your role and value to potential employers.
How to Get Into Clinical Research Without Experience?
To get into clinical research without experience, consider options like volunteering at a research institution, seeking out internships, taking relevant online courses, actively networking with professionals in the field, and highlighting transferable skills on your resume while applying for entry-level positions; this will allow you to gain practical experience and build your knowledge base to eventually pursue more advanced roles in clinical research.

Certify First: Career shifts starting with practical certifications like the CCRPS Training Program, which provides you with the theoretical and practical foundations to excel.
Focus Your Search on Adjacent Roles: Investigate roles like research assistantships or clinical trial supporting positions as an accessible path to gain field experience.
Volunteer at Research Facilities: Clinical research hubs in academic programs often list facilities needing data entry or support contributions, bypassing conventional job postings.
Lesser-Known Tips for Aspiring CRAs
Expand Regulatory Knowledge Globally: Going beyond U.S.-focused compliance? Certifications such as IAOCR Certification cover global exposure.
Master Technological Tools Early: Platforms for CRA Clinical Trials, like Medidata and Oracle CTMS, are becoming industry standards. Become fluent in these for an edge.
Specialize in Niche Fields: Gene therapy, AI-related trial compliance, and wearable tracker technologies dominate emerging roles.
Pro Tip: Engage in targeted training through CRA Training Institutes for niche exploration options.
Primary Responsibilities of a CRA
A Clinical Research Associate (CRA) is primarily responsible for overseeing and monitoring clinical trials at research sites, ensuring that studies are conducted according to the protocol, applicable regulations (like GCP), and ethical standards, by conducting regular site visits, reviewing data, identifying potential risks, and reporting findings to the sponsor company.

Clinical Trial Monitoring: Overseeing the progress of trials to ensure compliance with protocols and regulatory requirements, such as ICH-GCP. A CRA trainee program can help aspiring professionals gain expertise in this area.
Data Verification and Accuracy: Reviewing collected research data to ensure it aligns with trial objectives and has been recorded correctly. A CRA training program teaches these essential skills.
Regulatory Audits: Preparing reports for regulatory agencies and ensuring site readiness for inspections, an essential skill for those pursuing clinical research certifications.
Participant Safety Oversight: Safeguarding the rights, safety, and well-being of trial participants is a core responsibility of a clinical research associate.
Example: A CRA monitoring a COVID-19 vaccine trial ensures all data related to vaccination doses and observed side effects are meticulously recorded.
A CRA wears many hats—site monitor, data validator, compliance officer—and their work directly influences the success of clinical trials. If you’ve asked yourself what is a clinical research associate or what does a CRA do, understanding these core responsibilities is key.
Is This Career Right for You?
A CRA career, which stands for Clinical Research Associate, is likely a good fit for you if you have a strong interest in healthcare research, enjoy working with diverse teams at clinical trial sites, possess excellent communication and organizational skills, are detail-oriented, and are comfortable with frequent travel, as you will be responsible for monitoring and overseeing clinical trials at various locations. Becoming a CRA might be the perfect role for individuals who:
Have a background in life sciences, pharmacology, nursing, or health sciences.
Are interested in clinical research associate training to develop professional skills.
Possess strong organizational skills and attention to detail.
Are passionate about healthcare, technology, or pharmaceutical innovations.
Love a mix of office work and field visits, traveling to trial sites, and interacting with research professionals.
If you’re wondering about how to become a clinical research associate, we’ll cover that next.
How to Become a Clinical Research Associate? (Step by Step Guide)
To become a Clinical Research Associate (CRA), earn a health-related degree, gain clinical research experience, get certified for better job prospects, and network with industry professionals.

Step 1. Obtain the Right Education
The foundation of your CRA career begins with education.
Suggested Fields of Study:
Biology
Pharmacology
Nursing
Health Sciences
A bachelor’s degree is typically required, but advanced degrees, such as a Master’s in Clinical Research, can set you apart from other candidates. For those from non-health-related fields, certifications like those offered by CCRPS can close the knowledge gap.
Hidden Tip: Highlight any coursework related to clinical research or data analysis on your resume to demonstrate your understanding of the industry.
Step 2. Understand the Role of a Clinical Research Associate
Before you take the next steps, it’s essential to understand the CRA's responsibilities.
What Does a CRA Do?
Coordinate Clinical Trials: CRAs monitor multiple sites to ensure trials comply with Good Clinical Practice (GCP).
Verify Data Accuracy: They ensure the integrity of collected trial data.
Ensure Compliance: CRAs monitor regulatory adherence from study initiation to completion.
Participant Safety: They guarantee participant safety and well-being throughout the trial.
This knowledge is critical for tailoring your resume and cover letter to align with recruiters’ expectations.
Step 3. Earn Specialized Certifications
Certifications like CCRPS Clinical Research Associate Certification are globally recognized and demonstrate credibility to potential employers.
Recommended Certifications:
CCRA Certification (ideal for those with previous monitoring experience).
Clinical Trial Associate Certification (a great stepping stone for entry-level candidates).
IAOCR Certification (helps in international trial compliance).
Certifications not only validate your qualifications but also make your application stand out.
Step 4. Enroll in a CRA Training Program
A Clinical Research Associate Training Program bridges the knowledge gap between academic learning and hands-on experience. These programs cover essential areas like GCP, site monitoring, and patient safety protocols.
Programs like CCRPS also offer flexible, self-paced modules perfect for those balancing other commitments.
Step 5. Gain Entry-Level Experience in Clinical Research
Start your clinical research career with roles like Clinical Research Coordinator (CRC) or Clinical Trial Assistant (CTA). These positions provide valuable on-the-job experience that translates directly to CRA responsibilities.
Quick Tip: During interviews, talk about your experience handling patient recruitment, managing regulatory documents, or coordinating trial procedures. These are key skills CRAs need.
Step 6. Network with Industry Professionals
Leverage conferences, social networks like LinkedIn, and training program alumni networks to make connections in the clinical research field. Training programs such as CCRPS often facilitate networking opportunities. These connections can open doors to mentorships and job interviews.
Step 7. Tailor Your Resume for a CRA Position
Your resume is often the first impression employers have of you. Highlight the education, certifications, and experience most relevant to the CRA role.
Key Components of a Strong CRA Resume:
Professional Summary: A concise pitch of your goals and qualifications.
Example: “Certified Clinical Research Associate with experience in site monitoring, compliance oversight, and data validation seeks to contribute to impactful healthcare advancements within a dynamic research organization.”Education and Certifications: List degrees and certifications such as CCRPS Certification prominently.
Experience: Focus on measurable accomplishments in clinical research roles or related positions.
Example: “Achieved 100% audit compliance across trial sites, ensuring seamless regulatory reviews.”Skills Section: Include keywords such as trial monitoring, GCP compliance, and risk-based monitoring.
Pro Tip: Use action verbs like “monitored,” “coordinated,” and “validated” for impact.
Step 8. Craft a Winning Cover Letter
Your cover letter should be tailored to highlight your unique skills and passion for the field of clinical research.
Cover Letter Example:
Dear [Hiring Manager’s Name],
I am writing to express my interest in the Clinical Research Associate position at [Company Name]. As a certified CRA with hands-on experience in trial site monitoring and regulatory compliance, I am eager to contribute to your organization’s mission of advancing medical research.
Throughout my career, I have excelled in coordinating multi-site clinical trials, ensuring adherence to GCP standards, and fostering collaboration between sponsors and investigators. My recent completion of the CCRPS CRA Certification has further honed my expertise in trial management and patient safety protocols.
I am enthusiastic about applying my skills to ensure seamless trial coordination at [Company Name]. Thank you for considering my application.
Sincerely,
[Your Full Name]
Quick Tip: Research the company’s recent trials or major projects and reference them in your letter to show genuine interest.
Step 9. Apply for CRA Jobs
Armed with your tailored resume and cover letter, start applying for CRA roles. Look for opportunities with Contract Research Organizations (CROs), pharmaceutical companies, or academic medical centers.
Where to Apply:
LinkedIn
Glassdoor
Pharmaceutical and CRO-specific job boards
Company career pages
Consider starting with internships or part-time roles if you’re transitioning into clinical research.
Step 10. Enhance Industry Knowledge
To thrive as a CRA, stay updated on industry trends and regulations. Certifications like the CCRPS CRA Training Program focus on niche areas like decentralized trials and wearable technology monitoring.
Proactively learning about evolving areas of clinical research can propel you to senior roles or specialized opportunities.
Step 11. Prepare for Interviews
Acing the CRA interview requires a mix of technical expertise and strong communication skills.
Common Interview Questions:
“How do you ensure compliance with GCP guidelines in your monitoring process?”
“What challenges have you experienced during audits, and how did you resolve them?”
“How do you balance maintaining safety and achieving trial goals?”
Use examples to demonstrate your problem-solving skills and ability to work under pressure.
Step 12. Plan Your Career Growth
The CRA role is a stepping stone to various advanced career paths.
Career Pathways:
Senior CRA: Lead complex, multi-site trials.
Clinical Project Manager: Manage trial strategies and oversee budgets.
Regulatory Affairs Specialist: Focus on compliance policies and regulatory documentation.
Stay ahead by completing advanced certifications or enrolling in specialized courses through CCRPS to continually evolve in your career.
Salary Overview for Clinical Research Associates
A major incentive for becoming a CRA is the attractive compensation package. The salary for Clinical Research Associates varies depending on experience, certification, and geography.
Entry-Level CRA Salary
Newly trained CRAs can earn:
United States: $60,000–$75,000 annually.
Europe: €45,000–€60,000.
Australia/Asia: Salaries average $45,000–$55,000 USD.
Experienced CRA Salary
With 3–5 years of experience, earning potential jumps to:
United States: $85,000–$120,000 annually.
Senior CRAs: Can command salaries of $130,000 and higher.
Adding Certifications Boosts Pay
Certifications like CCRA certification or completing training through CCRPS can significantly increase earning potential.
Did You Know? Some organizations pay even higher for CRAs specializing in niche fields like oncology or rare diseases.
When discussing the potential salary of a CRA, remember that variables such as educational background, additional certifications, and job location also play a role.
A Day in the Life of a Clinical Research Associate
Ever wondered what a typical day looks like for a CRA? While the work can vary depending on the trial phase and employer, here’s a snapshot of common activities:
Morning: Reviewing updates from clinical trial sites and preparing monitoring reports.
Midday: Visiting clinical research sites to audit records, ensure protocol compliance, and address any site issues.
Afternoon: Meeting with site staff to provide training or resolve trial-related concerns.
Evening: Logging findings and preparing documentation to send to the sponsor or regulatory authorities.
Example: A CRA monitoring a psychiatric drug trial may need to confirm patient consent forms are stored accurately and that participant follow-ups are conducted on schedule.
This blend of diverse tasks keeps the role engaging and varied.
Common Challenges Faced by CRAs and Solutions
While being a CRA is rewarding, the role is not without challenges. Here’s how to overcome some common obstacles:
Problem: Managing multiple sites simultaneously.
Solution: Use organizational tools like spreadsheets and calendars to track visit schedules and updates efficiently.Problem: Addressing site staff resistance to trial protocols.
Solution: Build rapport and provide ongoing training to site staff, emphasizing how compliance ensures trial success and participant safety.Problem: Staying updated on changing regulations.
Solution: Enroll in continuous education programs such as the CCRPS CRA Certification to stay informed.
By understanding potential challenges and preparing to address them, CRAs can excel in their roles.
Networking Tips for Aspiring CRAs
Building a strong professional network is essential for success as a CRA. Here’s how to make industry connections:
Leverage LinkedIn: Join groups for CRAs or clinical research professionals, and engage in discussions.
Attend Industry Events: Conferences like DIA Global or BIO International are excellent for meeting sponsors and CRO representatives.
Utilize Training Networks: Programs like CCRPS often provide networking opportunities with alumni and industry leaders.
Quick Tip: Prepare an elevator pitch about your skills and career goals when meeting potential mentors or employers.
Your network can open doors to job postings, mentorship, and valuable advice.
Technological Tools Used by CRAs
With clinical trials becoming more digitally driven, CRAs must be proficient in various tools:
Clinical Trial Management Systems (CTMS): Track trial status, site progress, and compliance.
Electronic Data Capture (EDC): Handle and verify patient data through platforms like Medrio or Oracle Clinical.
Remote Monitoring Tools: Analyze patient data in real-time through wearable technologies or remote systems.
Example: A CRA working on decentralized trials may use Real-Time Data Sharing (RTDS) platforms to monitor patient vitals and ensure study compliance remotely.
Programs like the CCRPS CRA Certification often include modules on mastering these tools, giving you a career advantage.
Pharma vs. CROs: Which Is Best for You?
Choosing between working for a pharmaceutical company or a Contract Research Organization (CRO) depends on your career goals:
Pharma Companies:
Typically offer higher salaries.
CRAs oversee fewer trials but may have longer timelines.
Focus on in-house training and development.
CROs:
Provide exposure to diverse therapeutic areas and trial phases.
CRAs work on multiple concurrent projects, building experience faster.
Often lead to better global networking opportunities.
If gaining varied experience is your priority, CROs might be more suitable. Alternatively, pharma companies are ideal for those who want long-term trial involvement.
Regulatory Bodies and Their Roles
Knowledge of regulatory bodies is essential for CRAs to ensure compliance. Here are key organizations to be familiar with:
FDA (U.S.): Monitors drug and medical device trials to ensure ethical standards.
EMA (Europe): European counterpart of the FDA, overseeing EU trials.
ICH: Offers harmonized guidelines for Good Clinical Practice (GCP).
WHO (Global): Issues ethical and operational guidelines for multinational trials.
Fun Fact: A CRA working in America would need to be well-versed in FDA protocols, while those in Europe focus on FDA and EMA differences.
Understanding these organizations enhances your ability to monitor trials effectively within varying jurisdictions.
Ethical Considerations in Clinical Trials
CRAs are responsible for upholding strict ethical standards in clinical studies:
Ensuring Patient Rights: Confirm that participants fully understand and consent to the trials they are part of.
Preventing Conflict of Interest: CRAs must ensure sponsors do not influence findings.
Adhering to GCP Principles: Ethical treatment of patients is non-negotiable.
A CRA’s commitment to ethics significantly impacts the credibility of a clinical trial’s outcomes.
Example: Suppose a CRA discovers a site is underreporting side effects. It’s their duty to investigate and intervene, prioritizing participant safety.
Programs like the CCRPS CRA Certification emphasize ethics through practical modules and case studies.
The Importance of Soft Skills
Being a CRA is not just about technical knowledge—it also requires essential soft skills:
Communication: Effectively convey trial updates to site staff and sponsors.
Time Management: Balance multiple responsibilities, including site visits, documentation, and reporting.
Problem-Solving: Quickly address disruptions or errors during trials.
Quick Tip: If you’re transitioning into clinical research and worried about how to become clinical research associate-ready, soft skills are as critical as certifications.
These traits enable CRAs to thrive in high-pressure environments.
Career Transitions and Growth
For many professionals, becoming a CRA is a stepping stone to advanced roles:
Regulatory Affairs Specialist: Contribute to drug approval processes.
Senior CRA or Lead CRA: Guide and train junior CRAs while overseeing larger projects.
Project Manager: Manage entire clinical studies, from planning to closeout.
CRPs are increasingly bridging to AI-driven analytics roles post-certification enhancing usability adaptive monitoring practices.
Programs like CCRPS provide pathways for advancing within the industry, making career growth seamless.
Global Opportunities for CRAs
Clinical trials occur worldwide, creating immense potential for CRAs to work in diverse locations and cultural contexts.
Global opportunities include:
Decentralized Trials: Monitor trials conducted across multiple countries remotely.
Specialized Roles: CRAs with niche skills, like oncology or pediatrics, are in demand globally.
Regulatory Liaisons: Help international studies comply with varying national guidelines.
By understanding global dynamics, you can position yourself as a highly versatile candidate ready for international projects.
Pro Tip: Combine certifications like CCRPS CRA Training with knowledge of regulatory standards like ICH-GCP to thrive in global roles.
Take charge of your career and watch your role transform into one of the most impactful positions in modern healthcare. Your future as a CRA starts now.
Top Training Programs for CRAs (Based on Reviews and Results)
According to available information, some of the top training programs for Clinical Research Associates (CRAs) include: CCRPS (Clinical Research Professionals Society), which is often considered the leading option for comprehensive CRA training, Syneos Health's CRAI Foundation Training Program known for its structured approach, and Parexel's APEX CRA Program recognized for its strong industry reputation and focus on practical experience; with other notable options like the Mayo Clinic IATA Training for those working with biological samples in clinical trials.

1. CCRPS CRA Certification Training – Best Overall
Earned a reputation for its detailed modules, expert instruction, and hands-on approach. CCRPS allows students to:
Learn GCP principles, ethical considerations, and site monitoring techniques.
Access real-world case studies to prepare for industry challenges.
Benefit from job placement services to ease entry into the workforce.
2. IAOCR Certification Programs
Focused on international compliance, IAOCR is ideal for CRAs planning to work on multinational clinical trials.
3. IQVIA CRA Training
IQVIA stands out for integrating advanced data management and artificial intelligence into its curriculum.
4. Mayo Clinic IATA Training
This program emphasizes regulatory transport requirements for biological samples, catering to CRAs working in lab trials.
If you’re looking for the best CRA training programs, CCRPS consistently ranks at the top for flexibility and depth.
Frequently Asked Questions
What Qualifications Are Essential for Becoming a CRA?
Bachelor's degree in life sciences and certification from accredited programs like CCRPS.
How Long Does CRA Training Take?
Self-paced programs like CCRPS take 4–8 weeks, depending on the individual’s schedule.
What Is the Typical CRA Career Path?
Start as a Clinical Trial Assistant or Study Coordinator, gain certification, and transition to CRA roles.
Should I Get Certified?
Yes! Certifications like CCRA certification or a certificate from CCRPS make you stand out in job applications and provide essential skills for managing real-world trials.
Expert Tip: Include certifications in your LinkedIn profile to increase recruiter visibility.
What Is CRA Meaning in Medical Terminology?
CRA stands for Clinical Research Associate, a critical role in clinical trials ensuring compliance with protocols and regulations.
For anyone asking what is CRA certification and its benefits, certifications validate your ability to oversee scientific studies ethically and efficiently.
Advanced Opportunities for CRAs
CRAs have unique opportunities for growth and specialization, with career advancement options including:
CRA Team Manager: Overseeing a team of CRAs across multiple sites.
Clinical Project Manager: Managing entire clinical studies from planning to completion.
Regulatory Affairs Specialist: Moving into regulatory oversight roles for new trials.
CRAs specializing in newer fields like gene therapy or AI-integrated clinical trials are in especially high demand.
Example: A Senior CRA role for a multinational oncology trial can earn you $150K+ per year.
Why Choose CCRPS for CRA Training?
Choose CCRPS for CRA training because we offer a globally recognized and accredited certification, a comprehensive curriculum with real-world case studies, flexible online learning, expert instruction with mentorship, and a strong focus on career acceleration, making us a trusted option for aspiring Clinical Research Associates seeking to gain industry-relevant skills and secure jobs within the field.
Comprehensive training modules tailored to current industry needs.
Flexible self-paced learning for convenience.
Career advancement tools and networking opportunities to break into the industry.
Enrolling with CCRPS provides everything you need to become a confident, skilled CRA ready to tackle the challenges of 2025.
The pathway to becoming a Clinical Research Associate in 2025 is filled with opportunities to grow professionally and contribute meaningfully to global healthcare. With the right training, certifications, and experience, you could be monitoring innovative clinical trials that shape the future of medicine.
Take the first step by enrolling in the CCRPS CRA Training Program today—prepare for a rewarding and impactful career in clinical research.
Starting a career as a Clinical Research Associate (CRA) opens doors to medical innovation, competitive pay, and meaningful opportunities. With over 1.9 million science graduates each year, many face hurdles like entry-level roles requiring 1–2 years of experience or demanding lab work that falls short of career expectations. Clinical research training offers a fast-track solution.
Why Become a CRA?
CRAs ensure clinical trials are conducted ethically and efficiently, working alongside physicians and medical teams. Whether you’re aiming for experience before medical school or a new path entirely, CRA roles combine management and medical research into a dynamic, fulfilling career.
Key Benefits of a CRA Career
High Earnings: Certified CRAs earn $4,500–$12,000/month, with a 33% annual promotion rate.
Flexibility: Remote roles and opportunities to travel—expenses covered.
Career Growth: Start as a Clinical Trial Assistant or Coordinator and quickly advance with certification.
Get Ahead with CCRPS Training
CCRPS has 8 years of proven graduate success and resources institutions alone cannot provide due to our career and advanced education focus.
Fast-Track Timeline: Earn certification in as little as 4 weeks.
No Experience Needed: Our course prepares you for entry-level roles with no prior clinical experience required.
Expert Guidance: Learn from experienced professionals while building key industry connections.
Clinical Research Associate Certification: Your Path to a Rewarding Career
Are you a foreign doctor ready to make an impact in the U.S. healthcare system or seeking a high-growth, science-focused career? Becoming a Clinical Research Associate (CRA) could be your perfect solution.
Why Pursue a CRA Career?
Leverage Your Medical Expertise
Foreign doctors with medical degrees, such as MBBS, can bypass the challenges of USMLE and residency by transitioning into CRA or Clinical Research Coordinator (CRC) roles. Put your medical knowledge to work in a fulfilling alternative career.Gain a Distinct Skillset
CRA training provides expertise in conducting and managing clinical trials, adhering to ethical guidelines, and ensuring Good Clinical Practice (GCP). For a strong foundation, explore the ICH-GCP Course to master industry essentials.
The Most Comprehensive CRA Training with 196+ Modules
Our course is now even more robust, featuring over 196 modules—making it the most comprehensive CRA certification program available online. Designed by industry veterans, this curriculum ensures you're not just job-ready but set up for long-term success.
Path to Strategic Career Growth
General courses often fall short, leaving graduates unprepared. Our advanced training covers Senior CRA-level expertise, enabling you to secure coveted positions in top pharmaceutical companies, academic institutions, or research organizations. Interested in additional roles? Check out programs like the Clinical Trials Assistant Training for entry-level pathways or the Advanced Principal Investigator Certification for leadership opportunities.
Diverse Opportunities in a Thriving Industry
The clinical research field offers wide-ranging and flexible career options, from working with pharmaceutical giants like Pfizer to managing trials at prestigious academic institutions. Expand your knowledge in specialized areas through Pharmacovigilance Certification or Medical Monitoring Training to further enhance your career.

Clinical Research Associate Certification Qualifications
Foreign Doctors Welcome: A Clinical Research Associate or Coordinator plays a vital role in directing and supervising clinical trials conducted by physicians, nurses, and other science professionals. This career path is particularly attractive to many foreign doctors with completed medical degrees (MBBS) who can utilize their expertise in the US healthcare system by pursuing a CRA career instead of taking the USMLE or repeating residency training. For those interested in coordinating aspects, consider the Clinical Research Coordinator course.
Distinct Skillset: Unlike the traditional medical field you may be familiar with after years of schooling, Clinical Research Associate training provides a distinct and valuable skillset. For comprehensive understanding of Good Clinical Practice, see the ICH-GCP course.
Most Extensive Online Course: Our program goes beyond basic introductions, offering a comprehensive curriculum with over 110 modules – the most extensive Clinical Research Associate course available online. This in-depth training ensures you're well-prepared to secure a coveted CRA position.
Superior Coursework: Securing a CRA role is a strategic career move compared to the limitations of many traditional medical positions. While generic courses abound, we've observed that graduates often struggle due to a lack of substantive content. Our Clinical Research Associate course addresses this gap by providing Senior Clinical Research Associate-level training through 110 intensive modules grounded in the latest scientific principles. For those looking to assist in clinical trials, the Clinical Trials Assistant Training may also be of interest.
Diverse Career Opportunities: This high-demand science-based medical field offers diverse opportunities:
Work in the Private Sector: Pursue a CRA career with renowned pharmaceutical companies like Pfizer. Enhance your skills with the Advanced Clinical Research Project Manager Certification.
Academic Opportunities: Work in the academic sphere at medical schools. Those aiming for higher responsibilities may consider the Advanced Principal Investigator Physician Certification.
Unmatched Flexibility and Knowledge: In addition to our exceptional course content, we boast the largest number of clinical research courses available online, providing you with unmatched flexibility and knowledge. For those interested in safety monitoring of drugs, the Pharmacovigilance Certification and Medical Monitor Certification can enhance your capabilities in these critical areas.
Why Take A CRA Certification Course
Taking a CRA (Clinical Research Associate) certification course is beneficial because it demonstrates your expertise in clinical trial monitoring, regulatory compliance, and ethical research practices, which can significantly improve your job prospects in the field, open up career advancement opportunities, and give you a competitive edge when applying for CRA positions by validating your knowledge and skills to potential employers; even though certification isn't always mandatory, it is highly regarded within the industry.
Hidden Opportunities with Clinical Research Associate (CRA) Certification
A Clinical Research Associate (CRA) certification isn’t just about qualifying for a job—it’s about positioning yourself in one of the most dynamic and enriching career fields in healthcare. If you’re intrigued by clinical trials and medical innovation, here are some lesser-known but invaluable insights into the CRA certification process and its impact.
What Makes CRA Certification Unique?
Eligibility for Global Job Markets
A CRA certification opens doors not only in the U.S. but also in global markets. Clinical trials are conducted worldwide, and many pharmaceutical giants outsource research to international locations. Being certified with globally recognized credentials (like ACCRE) makes you competitive for roles in Europe, Asia, and beyond.Direct Contribution to Drug Safety
Did you know a CRA is instrumental in uncovering rare side effects of new treatments during trials? Their role in adverse event reporting ensures the safety of future drugs and devices. This critical function showcases how a CRA impacts healthcare on a global scale.Regulatory Expertise Across Borders
Certification programs don’t just teach you about Good Clinical Practice (GCP) guidelines—they prepare you to adapt to various regulatory frameworks like ICMRA standards or EU Clinical Trial Regulations, which vary from country to country.
Unrivaled Flexibility in Learning
Integrated Microlearning Modules
Certified programs now include bite-sized, module-based learning that accommodates even the busiest professionals. You can tackle complex topics in short, manageable segments, making learning less overwhelming without sacrificing depth.Simulated Trial Experiences
Some advanced CRA courses offer virtual clinical trial simulations. This allows you to experience trial management and monitoring in real-world scenarios, preparing you to hit the ground running.Personalized Career Development
Top-tier courses now include career mentorship programs, guiding students through networking, resume building, and interview strategies.
Jumpstart a Career Without Traditional Barriers
No U.S. Healthcare Background? No Problem
Many foreign professionals assume clinical research jobs demand U.S.-based experience. However, CRA certification provides all the foundational knowledge required to transition into the field and bypass requirements like USMLE or additional residency.Open to Non-Medical Professionals
While a healthcare or life sciences degree is beneficial, CRA certification is increasingly accessible for individuals with adjacent professional experience, like in project management or quality assurance. Some programs provide bridge courses for those without clinical backgrounds.Accelerate Your Career with Soft Skills
Beyond hard skills, certification ensures you develop essential soft skills like communication, data management, and attention to detail—critical factors often overlooked by non-certified applicants.
Lucrative and Less Explored Career Paths
Pharmaceutical Partnerships
CRAs can work directly with sponsors like Novartis or Amgen, managing trials that test cutting-edge therapies such as gene-editing drugs. These projects offer a front-row seat to medical breakthroughs.Remote CRA Roles
Few know that many CRA positions are 100% remote, cutting down the need for constant travel. Virtual monitoring of trials has become standard in many companies thanks to advanced technology.Niche Opportunities in Rare Disease Trials
Specializing in rare disease clinical trials connects you with groundbreaking projects where CRAs play a larger role due to the smaller teams involved. This niche often provides higher earning potential.
Behind-the-Scenes Secrets of CRA Certification
Specialized Certification Bonus
Many companies offer salary bonuses for obtaining specialized certifications alongside CRA credentials, such as Pharmacovigilance or Medical Monitoring.Direct Path to Leadership
Certification isn’t the endpoint; many certified CRAs transition into advanced roles like Clinical Research Managers or Project Leaders within three to five years.Boost Your Job Prospects
Even with general certification, including training from an ACCRE-accredited program, you'll join a network of former students, providing insights into hidden job openings and employer preferences.
Advanced Certification for Diverse Opportunities
Pairing CRA certification with additional qualifications expands your versatility:
Clinical Trial Auditing Certification lets you oversee trial compliance for top regulatory agencies like the FDA or EMA.
Oncology-Specific Training equips you to manage trials in one of the fastest-growing fields in medicine.
The Best CRA Certification Course for Entry-Level Professionals
Breaking into the field of clinical research as a Clinical Research Associate (CRA) can feel daunting, especially at the entry level. There’s a significant demand for well-trained CRAs, but companies are often hesitant to hire individuals without a strong background or certification. Why? The stakes are high—patient safety and trial integrity depend on skilled monitors. That’s where proper training becomes essential.
If you’re searching for a rigorous, results-driven CRA certification course that equips you with all the essential skills to excel, here’s everything you need to know.
Why Choose the CCRPS CRA Certification Course?
The CCRPS CRA Certification Course has been consistently recognized as one of the most comprehensive and effective clinical research programs online. Tailored for entry-level candidates, it bridges the experience gap with in-depth, hands-on learning that prepares students to hit the ground running.
What Sets This Course Apart?
Comprehensive Curriculum
Unlike many courses offering only 5-20 interactive modules, CCRPS provides over 196 densely packed modules covering every facet of clinical research monitoring. Topics include regulatory guidelines, protocol adherence, monitoring techniques, ethical considerations, and real-world scenarios you may encounter on the job.Advanced Training Beyond Basics
While other platforms focus on cursory introductions, CCRPS trains all students at a Senior CRA level, regardless of their prior experience. The material is designed to not only help you ace job interviews but also to excel in your role once hired.Expert-Led Instruction
The program is taught by seasoned industry professionals with over 15 years of hands-on CRA experience. They bring invaluable insights from the field, and students can ask questions privately after each session, ensuring personalized guidance.Guaranteed Preparedness
From learning how to interpret complex medical protocols to mastering compliance with regulatory standards, CCRPS equips you with the skills you need to succeed. Students are also trained to handle challenges specific to clinical research, such as managing adverse events and ensuring trial integrity.Success Rate You Can Trust
Currently, an impressive 82% of CCRPS graduates secure CRA positions within the first month of completing the course.Optional Internship Experience
For candidates with limited experience or those looking to strengthen their resumes, CCRPS offers a 6-month remote internship opportunity. This hands-on training ensures that you gain real-world exposure while actively applying for jobs.Flexible Formats
The program is fully online, making it accessible for professionals with busy schedules. Despite its remote format, the course fosters interaction through discussion forums, mentorship, and a curriculum designed for immersive online learning.
Who Can Take the Course?
The course welcomes a diverse range of professionals, including science graduates, nurses, and even physicians looking to transition to clinical research. No prior CRA experience? No problem. The course is specifically designed to train and certify individuals from various backgrounds, ensuring that everyone receives the same advanced-level education.
Unique Insights Into CRA Certification
Medical Knowledge Isn’t Enough
Although medical professionals often transition into CRA roles, medical knowledge alone won’t suffice. Clinical research monitoring requires a deep understanding of protocols, guidelines, and ethical considerations, which this training focuses on.Standout Feature
The program includes all potential scenarios you might face as a CRA, arming you with the tools to handle even the most complex trials.Industry-Recognized Accreditation
CCRPS is ACCRE-accredited, a hallmark of quality and industry recognition.
Career Advice and Job Placement Resources
After certification, the next step is securing a position. While CCRPS does not directly place students in jobs, it offers invaluable guidance and resources to help you succeed in the job market.
Professional Resume & Cover Letter Support
TopCV: Upload your resume for a free review and feedback that improves your chances of standing out.
ResumeRabbit: Distribute your resume to over 60 job boards, saving you time and increasing visibility.
Recruiting & Job Boards
Clinical Trial Recruiters via I-Recruit: Access specialized recruiters who work with candidates in clinical research.
Indeed: A go-to platform for up-to-date job bulletins in clinical research and related fields.
Clinical Research Guidance
Clinical Trial Podcast with Kunal: Provides insights into the clinical research industry and advice for navigating your career.
Networking Opportunities
Engage with online CRA communities or LinkedIn groups dedicated to clinical research. Many jobs aren’t advertised publicly, so networking can open doors to hidden opportunities.
Application Tips for CRA Jobs
Tailor Your Applications
Always customize your CV and cover letter for each specific role and company. Use the job description as a guide to highlight relevant skills and experiences.Follow Up
If you don’t hear back, don’t assume the application was rejected—it may have been overlooked. Follow up to ensure your application gets the attention it deserves.Showcase Your Certification
Mention your CCRPS certification prominently, detailing key skills you gained from the course that align with the job requirements.Highlight Internship Experience
If you’ve taken advantage of CCRPS’s internship, list your responsibilities and achievements to demonstrate hands-on experience.
Resume Writing Services
ResumeGo - Offers industry-specific, keyword-optimized resumes with one-on-one consultations and unlimited revisions. Learn more.
Let’s Eat, Grandma - Provides tailored resume writing services with a focus on detailed consultations.
TopResume - Known for its free resume review and emphasis on keyword optimization. Check it out.
Job Boards for Clinical Research
PharmiWeb.Jobs - A comprehensive job board for clinical research roles, including Clinical Research Associate positions. Visit PharmiWeb.
HCSRN Job Board - Lists faculty and staff positions at research centers and academic institutions. Explore HCSRN.
Networking Opportunities
LinkedIn Groups - Join groups like "Clinical Research Professionals" to connect with peers and discover job opportunities.
Clinical Trial Podcast - Offers insights and advice for navigating the clinical research industry. Listen here.
Exploring the ICH-GCP in Clinical Research
Clinical research is more than just testing new treatments—it's about ensuring the safety and dignity of participants while generating data that is trustworthy and accurate. To achieve this, clinical studies globally adhere to the ICH-GCP guidelines—the International Committee for Harmonization of Good Clinical Practice. These guidelines set the gold standard for safeguarding human participants' rights and integrity while maintaining the validity of the research process.
Why are these standards so important? For one, they ensure clinical trials respect the rights and dignity of every patient involved. Secondly, they outline how data should be collected, securely stored, and analyzed to ensure accuracy. Regulatory bodies, such as the FDA in the U.S., have enforced compliance with these guidelines, including detailed protocols like the FDA’s E6(R2) Guidance for Good Clinical Practice.
Sticking to these international standards isn’t just good practice—it’s essential for anyone pursuing a career in clinical research. If you’re aspiring to make your mark as a Clinical Research Associate (CRA), understanding the ICH-GCP is your starting point.
What Does It Take to Become a CRA?
A CRA’s role involves monitoring clinical trials, ensuring proper protocol adherence, and safeguarding participant safety. But what skills and qualifications do you need to thrive in this career path?
Educational Foundation
A degree in a biological science, medicine, nursing, or life sciences is commonly required. Degrees in fields like pharmacology, biochemistry, or immunology are particularly valuable.Technical Expertise
CRAs are expected to manage essential data systems like Electronic Data Capture (EDC), handle regulatory documents, and analyze clinical trial data.People Skills
Equally crucial is emotional intelligence. Strong interpersonal skills are non-negotiable since CRAs often coordinate between various teams, troubleshoot issues, and guide study sites towards compliance.
Beyond technical and interpersonal expertise, being detail-oriented and possessing a thorough understanding of medical trial regulations are vital traits. The ability to spot compliance violations and implement corrective actions ensures the success and integrity of the study.
Build Your Career Path as a CRA
Gone are the days when medical or nursing professionals could transition into CRA roles without research training. Today, most employers seek individuals with direct clinical research experience or specialized training. While Clinical Trial Assistants (CTAs) and Clinical Research Coordinators (CRCs) often receive preference, aspiring CRAs now have access to professional courses that open career doors.
For instance, master's programs in clinical research offered by universities are a solid option but can take years to complete. Those who want a faster yet comprehensive route can explore certification programs designed specifically for CRAs—programs like the Advanced Clinical Research Associate Certification (ACRAC).
A Toe in the Door for Non-Clinical Professionals
Think you can’t become a CRA without prior experience or a specialized degree? Think again.
The CCRPS ACRAC program is one of the most accessible courses for candidates without a clinical research background. What makes it unique?
Comprehensive Curriculum
With over 196 high-impact modules, the program dives deep into every possible scenario you could face as a CRA. The training equips you to master regulatory requirements, research ethics, trial protocols, and compliance standards.Accessible to Fresh Graduates
You don’t need years of lab work or experience as a Clinical Research Coordinator. With just a B.S. degree in a life science, this program prepares you to step directly into a CRA role.Hands-On Guidance
Taught by experts with 15+ years in the field, the program offers personalized support and mentorship, helping you polish your skills and readiness for job interviews.Industry Accreditation
The ACRAC program carries multi-accreditation from respected institutions like ACCRE, ACCME, and ANCC, giving your certification global recognition.Optional Remote Internship
To boost your resume, CCRPS even offers an internship option, allowing you to gain hands-on experience while applying for jobs.
These opportunities make the ACRAC program an unparalleled option for breaking into clinical research.
Your Next Step
A career as a Clinical Research Associate offers not only competitive salaries but a chance to directly contribute to life-saving medical innovations. With the growing demand for CRAs and the right training, you can kickstart your career without waiting for years of experience.
Take the leap today. Enroll in the Advanced Clinical Research Associate Certification (ACRAC) and ensure your place in this exciting, impactful field.
Elevate Your Career in Clinical Research with CCRPS ACRAC
If you’ve been dreaming of becoming a Clinical Research Associate (CRA) or want to take your clinical research career to the next level, the CCRPS Advanced Clinical Research Associate Certification (ACRAC) is the definitive program for comprehensive, hands-on training. This meticulously structured program doesn’t just set the gold standard—it ensures you’re fully prepared for one of the most rewarding, impactful careers in healthcare and research.
Here’s an in-depth look at how the ACRAC program sets you up for success.
Flexible and Achievable Learning
Studying for a new career shouldn’t mean putting your current life on hold. With the ACRAC program’s self-paced structure, you have the freedom to choose how and when you study. The program offers approximately 250 hours of in-depth training content, spread across 196 modules that build your competence step-by-step.
For students dedicating full-time focus to their studies, certification can be achieved in just two to three weeks. But for those balancing other responsibilities, you can tailor the program to your rhythm and study at your own pace. This flexibility ensures access to a robust educational foundation without unnecessary pressure.
An Exhaustive Curriculum Covering Every CRA Competency
The breadth and depth of the curriculum are unmatched. With 196 detailed modules, the ACRAC program ensures you acquire expertise across every key area of clinical trials and research monitoring. Here’s how the training is structured:
Core Foundations
Introduction to clinical research terminology, concepts, and abbreviations.
Overview of the principles of Good Clinical Practice (GCP), informed by both ICH and FDA guidelines.
Understanding the regulatory framework and compliance obligations, including FDA E6(R2) guidelines and key CFR sections.
Ethics and Compliance
Comprehensive training in designing trials that meet ethical standards for human subjects, particularly for vulnerable populations such as children, pregnant women, and individuals with impaired consent capacity.
Understanding informed consent procedures, minimizing coercion, and maintaining transparency with trial participants.
Clinical Trial Protocol Development
Step-by-step guidance on the creation, implementation, and documentation of Clinical Trial Protocols (CTPs) in compliance with ICH-GCP standards and federal regulations.
Training in logistical essentials, such as IRB/IEC approval processes and navigating trial budgeting and funding.
Pharmacovigilance and Adverse Event Management
Instruction on identifying, documenting, and managing Adverse Events (AEs), with distinctions between different categories like Adverse Drug Reactions (ADRs) and Serious Adverse Events (SAEs).
Mastering causality assessments for adverse events and ensuring clear communication with IRBs and sponsors.
Site Visits and Trial Monitoring
Thorough preparation for all site visit types, including:
Site Qualification Visits (SQVs) for assessing site readiness and investigator qualifications.
Site Initiation Visits (SIVs) for introducing site staff to trial protocols and regulatory expectations.
Routine Monitoring Visits (RMVs) for supervising ongoing trial compliance.
Close-Out Visits (COVs) to ensure all documentation and compliance tasks are complete post-trial.
Extensive focus on documentation, including Case Report Forms (CRFs), Trial Master Files (TMFs), and Source Data Verification (SDV).
Tools and Technology
Mastery of digital tools used in modern clinical trials, such as Electronic Data Capture (EDC) systems, Interactive Response Technologies (IRT), and remote monitoring software.
Training in adopting new and evolving technologies, including risk-based monitoring (RBM) and centralized monitoring practices.
With each module, you’ll build a clear understanding of both the theoretical foundations and the practical application of clinical trial protocols. The program’s thoughtful design ensures emerging CRAs are equipped to seamlessly integrate into the workforce.
Real-Life Problem Solving for CRAs
The ACRAC program goes beyond theoretical training. Through realistic scenarios and case studies, you’ll learn how to handle critical challenges that CRAs face on the job, such as:
Identifying protocol deviations and violations, escalating when necessary, and working with study teams to implement corrective actions.
Dealing with ethical misconduct or research fraud without compromising the integrity of trials.
Maintaining the delicate balance of safeguarding participants’ rights and ensuring smooth trial progress.
These hands-on exercises ensure you’re ready to tackle unforeseen challenges, making you a valuable asset to any clinical research team.
CME Credits for Career Growth
The ACRAC program doesn’t just certify you—it enhances your professional credentials. It provides 17.5 Continuing Medical Education (CME) credits, recognized by prestigious institutions like the Accreditation Council for Clinical Research & Education (ACCRE). These credits can boost your resume and help you build expertise in other healthcare-related industries like medicine, nursing, or pharmacy.
Why ACRAC is Ideal for Aspiring CRAs?
Whether you’re fresh to the field or pivoting to clinical research from another career, ACRAC is tailored to meet your unique needs. Unlike many programs, it assumes no prior experience in clinical research. Instead, it focuses on teaching you everything you need to know to perform at the level of a senior CRA.
With certifications from globally respected bodies like ACCME and Transcelerate Biopharma, the program ensures your qualifications are recognized internationally, opening a world of job opportunities.
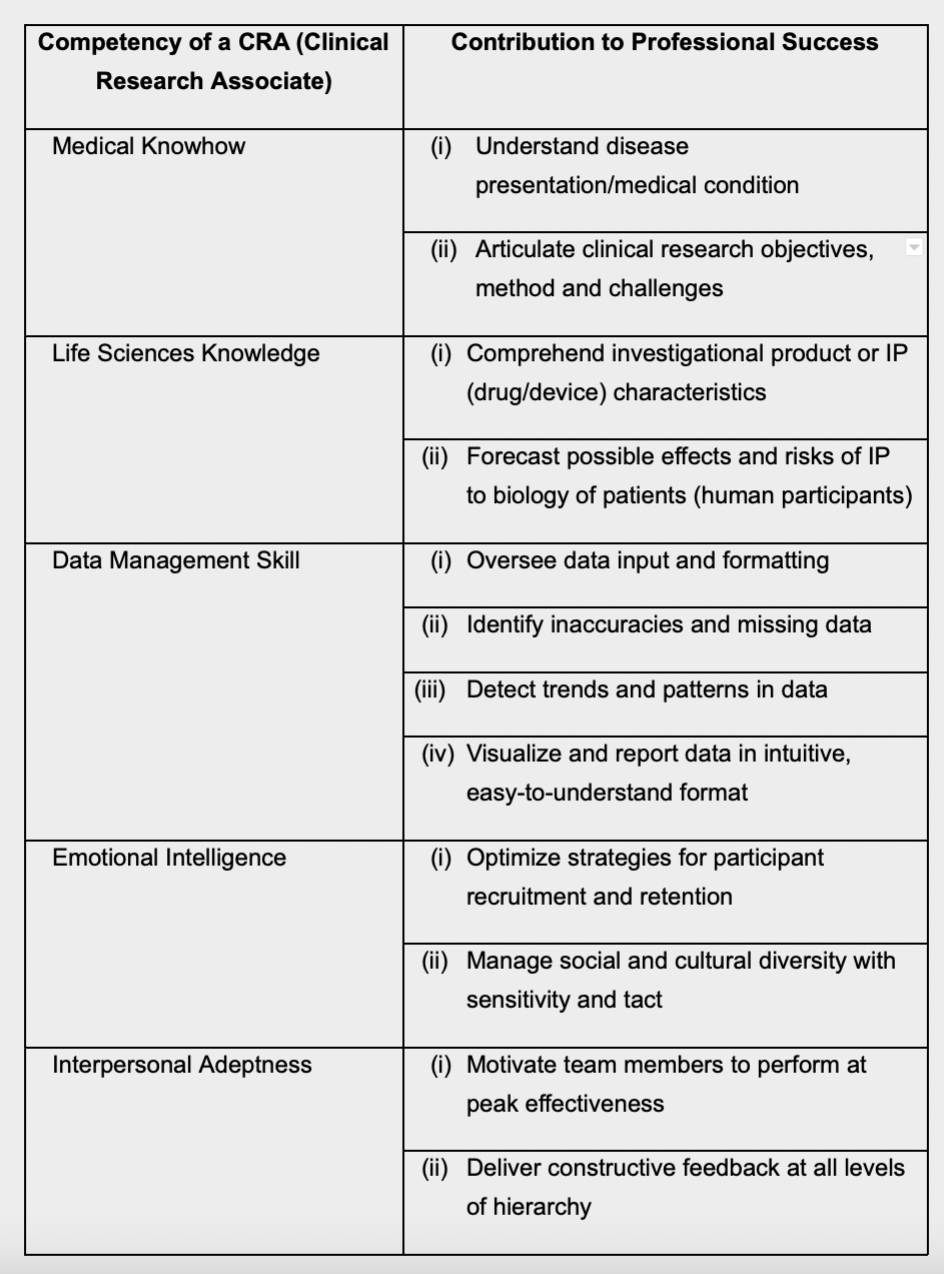
Core Competency Framework for CRAs
To illustrate, the ACRP’s ‘Core Competency
Framework for Clinical Study Monitoring’
requires that a CRA should be able to identify
and correct compliance violations at a study
site. The CRA must not only bring such
violations to the attention of site staff, s/he
must induce them to take corrective action,
as well as reporting the matter and even
escalating it, where necessary14.
The table below summarizes the ideal
competencies of a CRA, and provides
insights on how each ability contributes to
the CRA’s performance.

A Comprehensive Look at the ACRAC Syllabus for Clinical Research Associate Training
The CCRPS Advanced Clinical Research Associate Certification (ACRAC) syllabus is an exceptional, all-encompassing program meticulously crafted to provide aspiring Clinical Research Associates (CRAs) with the knowledge, expertise, and tools to thrive in the clinical research industry. It’s an extensive syllabus, not just a training program—it’s a launching pad for an impactful career in clinical research.
With 196 thoughtfully curated modules, the ACRAC program dives deep into every competency and skill required for CRAs at every stage of their career. Whether you’re new to the field or looking to enhance your expertise, this syllabus ensures you’re fully prepared to monitor and manage clinical trials with confidence and professionalism.
Foundations to Advanced Clinical Monitoring
The program kicks off with a solid foundation in clinical research essentials. Through the Introduction section, you’ll get acquainted with the critical roles and responsibilities of a CRA, the principles of Good Clinical Practice (GCP), and how ethical and legal standards govern the clinical trial landscape.
Stakeholder Collaboration: You’ll explore how CRAs collaborate with sponsors, IRBs, investigators, and site staff to ensure harmony between regulatory requirements and trial objectives.
ICH-GCP Overview: This part unpacks global guidelines, like the ICH GCP and FDA’s standards, crucial for the smooth execution of clinical trials, including a forward-looking discussion on trends in monitoring.
Sponsor and Investigator Roles
Moving into the Sponsor and Investigator Roles modules, the syllabus deepens your understanding of the complex partnerships in clinical research. Covering key sections of ICH GCP (E6), you’ll learn about reporting responsibilities and the critical measures sponsors and investigators must take to maintain participant safety, data validity, and compliance.
These modules are particularly valuable for CRAs tasked with maintaining open communication between parties and ensuring no stone is left unturned in fulfilling regulatory expectations.
Clinical Trial Phases and Procedural Expertise
The program exhaustively reviews all phases of clinical trials—from preclinical to post-marketing studies—with a focus on monitoring techniques tailored to each phase’s unique demands. Highlights include:
Safety Compliance: Learn best practices for ensuring trial safety and managing unexpected challenges during monitoring visits.
Trial Design Techniques: Mastering protocol-specific methodologies to ensure data accuracy and participant welfare.
Specialized Trials: You’ll gain insights into handling trials involving vulnerable populations, including children and individuals with cognitive disabilities. These modules prepare you to manage ethically sensitive scenarios with confidence.
Advanced Clinical Trial Protocol Development
The importance of a well-crafted Clinical Trial Protocol (CTP) cannot be overstated. The syllabus dedicates an entire section to guiding you through the drafting, reviewing, and assessing process. You’ll learn how to define inclusion and exclusion criteria while addressing common complexities, such as trials involving older adults or pregnant women.
Protocol Deviations and Violations: Handling major and minor deviations is a skill every CRA needs. You’ll learn how to identify, mitigate, and resolve these issues effectively.
Site Monitoring Visits
CRAs are the backbone of site monitoring, a responsibility that requires impeccable preparedness and communication. The syllabus provides unparalleled training on all types of monitoring visits:
Site Qualification Visits (SQVs): Get step-by-step guidance for pre-study checks, investigator selection, and evaluation.
Site Initiation Visits (SIVs): Learn how to prepare documentation, educate site staff, and communicate protocols to ensure a strong trial launch.
Routine Monitoring Visits (RMVs): Gain expertise in ensuring compliance, tracking progress, and correcting deficiencies during active trials.
Site Close-Out Visits (COVs): Equip yourself with tools to confirm proper data submission, secure records, and leave no loose ends upon trial completion.
Practical templates and checklists accompany these modules, so you hit the ground running from day one.
Modernized Monitoring Strategies
Keeping up with advancements in technology is a major component of the ACRAC program. Tackling everything from electronic data capture (EDC) systems to risk-based monitoring (RBM) approaches, you’ll learn how modern tools enhance trial efficiency and data accuracy.
Specialized modules on remote and centralized monitoring, which became especially critical during the COVID-19 shift, ensure you’re up to date with the latest methods.
Pharmacovigilance and Regulatory Affairs
Safety is at the heart of clinical research, and the program’s focus on pharmacovigilance ensures you’re prepared to monitor adverse events (AEs) and maintain compliance with regulatory requirements.
Adverse Events Reporting: From causality analysis to periodic updates, you’ll master the protocols for identifying, documenting, and reporting safety concerns.
Investigational Products: Learn storage, inventory, and accountability best practices for medical products in a way that ensures full traceability.
Specialized Monitoring Techniques
The syllabus dives into niche areas of monitoring, including oncology, gene therapy, and rare disease trials. These specialized modules will prepare you to adapt to evolving demands of unique trial types and patient populations.
Leadership, Team Management, and Advanced Strategies
Leadership and decision-making skills are integral to a CRA's role. The syllabus equips you with strategies to manage cross-functional teams, negotiate with sponsors, and build collaborative relationships with stakeholders.
The advanced strategy modules elevate your skills further, offering insights into modern methodologies like adaptive monitoring, predictive analytics, and machine learning applications in trial oversight. These cutting-edge tools are essential for data-rich environments in global trials.
Patient Recruitment, Retention, and Misconduct Prevention
From bolstering patient engagement to navigating ethical dilemmas, these modules ensure you’re well-versed in managing participant interactions. Learn how to identify motivating factors for recruitment, address compliance challenges, and recognize red flags for misconduct or fraud.
Regulatory Mastery with TMFs and CFR 21 Part 11
Managing Trial Master Files (TMFs), essential regulatory documents, and electronic signatures per CFR 21 Part 11 is simplified with these practical modules. The emphasis on data accuracy and reliability ensures that no detail is overlooked.
Final Preparations and Certification
The program concludes with real-world exercises and an exhaustive evaluation through case studies and a certification exam. By the end, you’ll be fully equipped to excel in advanced monitoring practices and meet the demands of a senior-level CRA.
Why Choose the ACRAC Syllabus?
The ACRAC syllabus sets itself apart with its depth, versatility, and applicability. With practical insights, global relevance, and accreditation from esteemed institutions, it’s not just a certification—it’s a career-transforming experience.
Start Your Journey Today
Whether you’re beginning your career or looking to level up, the ACRAC program gives you the tools and training to thrive in clinical research. Explore the program and enroll now at CCRPS ACRAC to start advancing your skills. With these 196 modules behind you, success is just around the corner!

Clinical Research Associate Training
Get ahead in clinical research with advanced accredited online CRA certification for $450. Demo our on-demand course below.
Clinical Research Associate Certification
References
Beroe Inc. - Explore the latest insights and market intelligence on clinical research organizations, focusing on trends, challenges, and opportunities in the industry. Visit Beroe Inc.LinkedIn Jobs - Discover entry-level Clinical Research Associate positions across the United States, leveraging your network to find new opportunities. Explore LinkedIn Jobs
CenterWatch - Read about the ongoing shortage of qualified CRAs and its impact on the pharmaceutical industry, including increased costs and project delays. Read more on CenterWatch
NCBI - Access a comprehensive article on clinical research, providing valuable insights into methodologies and best practices. Read the article on NCBI
NIAID - Learn about the DMID's role in investigational product research, including guidelines and protocols. Visit NIAID
FDA - Understand the different types of clinical research and their significance in advancing medical knowledge. Explore FDA's guide
Dixon JR. 1999 - Review the international conference on harmonization good clinical practice guideline, focusing on quality assurance in clinical trials. DOI: 10.1080/105294199277860
FDA E6(R2) Guidelines - Review the comprehensive guidelines for Good Clinical Practice, ensuring compliance and ethical standards in clinical trials. Access the guidelines
WHO - Discover the recommended format for research protocols as outlined by the WHO's Research Ethics Review Committee. Visit WHO
IAOCR - Find guidance on securing your first clinical research job, with tips and strategies for breaking into the field. Explore IAOCR
New Scientist - Gain insights into building a career in clinical research, including necessary skills and industry trends. Read more on New Scientist
NCBI - Delve into data management practices in clinical trials, focusing on efficiency and accuracy. Read the article on NCBI
St. Germain DC, Good MJ. 2017 - Explore data management in clinical trials, as detailed in the book "Principles and Practice of Clinical Research." ISBN 978-0-12-849905-4
ClinEssentials - Offers a variety of courses, products, and services designed to help clinical research professionals thrive and achieve fulfilling careers. Visit ClinEssentials
Clinical Leader - Explore insights and advice for starting a career in clinical research, including common challenges and solutions. Read more on Clinical Leader
ProClinical - Learn how to secure a job as a Clinical Research Associate, with tips on standing out in the application process. Visit ProClinical
SWOG Clinical Research Resources - Provides a comprehensive collection of links to useful resources related to clinical trials, including those from the FDA, OHRP, OCR, NIH, and NCI. Explore SWOG Resources
College Choice - Review the best master's degree programs in clinical research, helping you choose the right path for advanced education. Visit College Choice
ClinEssentials - Offers a variety of courses, products, and services designed to help clinical research professionals thrive and achieve fulfilling careers. Visit ClinEssentials
SWOG Clinical Research Resources - Provides a comprehensive collection of links to useful resources related to clinical trials, including those from the FDA, OHRP, OCR, NIH, and NCI. Explore SWOG Resources