
Informed Consent: What Every Clinical Researcher Must Know
In clinical research, informed consent is more than just a procedural requirement—it is the ethical foundation of participant engagement. It ensures individuals understand the purpose, risks, and benefits of a study before agreeing to participate. This process safeguards both the rights of participants and the integrity of the research itself.Informed consent is not about paperwork; it is about clear communication. Researchers must provide information in a manner that participants can comprehend, ensuring they make an informed decision without coercion or undue influence.
A failure to obtain proper informed consent can lead to regulatory penalties, compromised research integrity, and potential harm to participants. Regulatory bodies like the FDA, EMA, and IRBs enforce strict guidelines to ensure compliance. With increasingly complex study protocols, clinical researchers must remain vigilant in maintaining ethical and legal standards. Informed consent is not optional—it is a core component of ethical research conduct and reflects a commitment to human dignity and rights.

Fundamentals of Informed Consent
Definition and Importance
Informed consent in clinical research is the participant’s explicit agreement to partake in a study, provided they have received full and understandable information about the study’s purpose, procedures, risks, benefits, and alternatives. This principle upholds autonomy, allowing participants to make informed choices free from coercion. Consent isn’t a one-time event but an ongoing process throughout the research.
Clear communication is essential to prevent misunderstandings or uninformed decisions. Researchers must provide comprehensive information tailored to the participant’s language and comprehension level. This includes explaining risks, benefits, data protection measures, and the right to withdraw at any time. Informed consent enhances the researcher-participant relationship, fostering trust and transparency. It protects institutions and sponsors from legal risks and reputational damage by ensuring compliance with FDA regulations, ICH-GCP standards, and local ethics requirements.
Legal and Ethical Framework
The legal foundation of informed consent comes from global documents like the Declaration of Helsinki, the Belmont Report, and local laws such as the U.S. Code of Federal Regulations (CFR) and Europe’s Clinical Trials Regulation (CTR). These rules mandate that consent is informed, voluntary, and well-documented.
Ethically, it embodies principles of autonomy, beneficence, and justice, ensuring participant rights are respected, risks minimized, and benefits maximized. Special protections apply to vulnerable populations—such as minors, cognitively impaired individuals, or economically disadvantaged groups. Researchers must secure assent from minors or proxy consent where applicable. Oversight bodies like Institutional Review Boards (IRBs) and Ethics Committees (ECs) ensure that the consent process complies with legal and ethical standards. These bodies review consent documents, monitor processes, and audit records to protect participants and uphold transparency.
| Regulatory Body | Key Guideline | Core Focus |
|---|---|---|
| FDA | 21 CFR Part 50 | Protection of human subjects in clinical trials |
| EMA | Clinical Trials Regulation (EU CTR) | Harmonized standards for EU clinical research |
| ICH-GCP | E6(R2) Guidelines | Global good clinical practice standards |
| Declaration of Helsinki | World Medical Association’s Ethical Principles | Foundational ethics for human subject research |
Components of a Valid Informed Consent
Information Disclosure
For informed consent to be valid in clinical research, participants must receive comprehensive, clear, and relevant information about the study. This includes the purpose of the study, the procedures involved, potential risks and benefits, alternative treatments, confidentiality assurances, and the right to withdraw at any time. Information should be conveyed in simple, non-technical language that the participant can easily understand, tailored to their cultural and educational background.
Additionally, disclosure must cover the researcher's affiliation, funding sources, potential conflicts of interest, and how participant data will be stored and used. Consent materials should be updated promptly if the study protocol changes or new information emerges. Failure to provide complete and accurate disclosure compromises the participant’s ability to make an informed choice.
Comprehension Verification
Researchers must ensure that participants truly understand the information provided. This involves more than just giving them written materials to read and sign. Comprehension can be verified through interactive discussions, teach-back methods, and opportunities for participants to ask questions and receive clear answers.
Researchers should also assess for language barriers, literacy levels, or cultural factors that may impact comprehension. Informed consent is only valid when participants fully grasp the nature of the research and the implications of their participation.
Voluntariness
A cornerstone of valid informed consent is voluntariness. Participants must make their decision to enroll in a study freely, without coercion, undue influence, or misrepresentation. Researchers must avoid applying pressure, whether explicit or subtle, and must clearly inform participants that refusal to participate or withdrawal from the study will not result in penalty or loss of benefits.
Special care must be taken when involving vulnerable populations—including minors, cognitively impaired individuals, or economically disadvantaged groups. These populations may be more susceptible to undue influence, and additional safeguards must be implemented to protect their rights.
Poll: Do You Think Informed Consent is Well Understood by Clinical Researchers?
Thank you for submitting your response!
Process of Obtaining Informed Consent
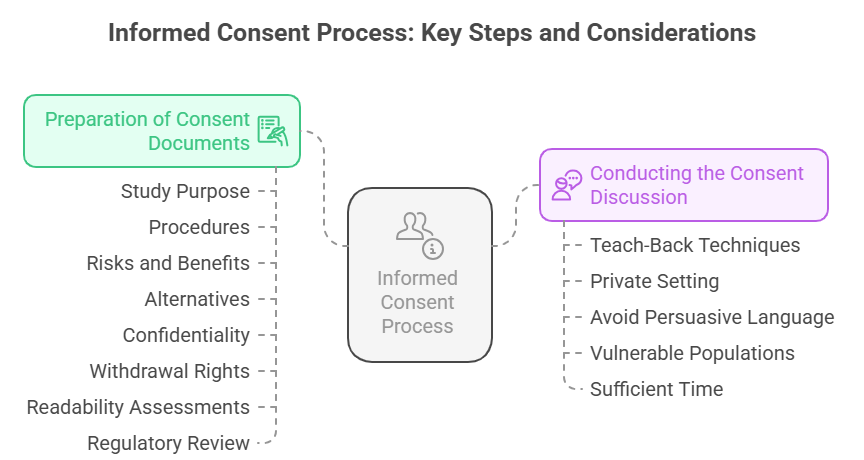
Preparation of Consent Documents
Informed consent begins with the development of clear, concise, and participant-friendly documents. Consent forms must explain the study’s purpose, procedures, potential risks and benefits, alternatives, confidentiality, and the right to withdraw. These forms should use simple language, avoiding technical jargon, and must be culturally and linguistically appropriate. To prepare effective consent documents, researchers should conduct readability assessments, consult community representatives if the study involves diverse populations, and pretest the forms for comprehension.
Regulatory bodies such as IRBs or Ethics Committees review and approve consent documents to ensure they meet legal and ethical standards. Content clarity is non-negotiable. Participants must not only read but also fully comprehend what they are agreeing to. Forms must highlight key information and include sections for participant questions, ensuring they have every opportunity to seek clarification before signing.
Conducting the Consent Discussion
The consent discussion is an interactive exchange between the researcher and the potential participant. It is not merely handing over a document to be signed but involves clear, two-way communication. Researchers must walk participants through the study details, addressing questions and verifying understanding through teach-back techniques, where participants explain what they understand in their own words. This discussion should occur in a private setting, free from distractions or pressure, to facilitate openness. Researchers should avoid persuasive language or any suggestion that participation is mandatory.
In cases involving vulnerable populations, extra care is needed to ensure comprehension and voluntariness, potentially involving a neutral third party or legal representative. Timing is essential. Participants must be given adequate time to consider participation, discuss with family or advisors, and make an informed decision. There is no ethical basis for rushing this process.
Challenges and Considerations
Language and Cultural Barriers
One of the most persistent challenges in obtaining valid informed consent is the presence of language and cultural barriers. In clinical research, participant populations are often diverse, with varying levels of literacy and different cultural norms. Researchers must ensure that consent forms and discussions are linguistically appropriate and culturally sensitive. Simply translating a form into a participant's native language is not enough. Effective consent involves using simple, culturally relevant language and engaging interpreters or cultural mediators when necessary.
Moreover, cultural norms may affect how participants perceive authority figures, risk disclosure, and decision-making autonomy. In some cultures, family or community leaders play a significant role in decisions, and researchers must balance respect for these norms with the ethical imperative of individual consent. Failure to address these barriers can result in participants consenting without truly understanding the study’s scope and implications.
Vulnerable Populations
Vulnerable populations, such as minors, cognitively impaired individuals, prisoners, economically disadvantaged persons, and others, require heightened protections during the consent process. These groups may have limited capacity to provide fully informed and voluntary consent. Researchers must implement additional safeguards to ensure their autonomy and rights are protected. For minors, this often involves obtaining both parental permission and the child’s assent, ensuring that the minor understands the study and agrees to participate.
In the case of cognitively impaired individuals, legal representatives or guardians may need to provide consent, and researchers should make every effort to involve the participant in the discussion as much as possible. The concept of voluntariness is particularly critical for vulnerable populations, as these groups may be more susceptible to coercion or undue influence. Researchers must take care to avoid any real or perceived pressure that might compromise the participant’s freedom to decide. Special ethical and regulatory considerations apply to these populations, including additional IRB/EC oversight, stricter documentation requirements, and tailored consent materials.
How CCRPS Certification Equips You to Handle Informed Consent with Confidence and Compliance
Informed consent is a foundational requirement in every clinical trial—and one of the most scrutinized elements during audits and inspections. CCRPS’s clinical research certifications are designed to give you a deep, working knowledge of how informed consent operates under ICH-GCP, FDA 21 CFR Part 50, and international ethical standards. Whether you're pursuing the CRA, CRC, PI, or GCP certification, informed consent is not treated as theory—it’s treated as a skill you must master.
Through scenario-based modules and compliance-focused lessons, CCRPS trains clinical researchers to manage informed consent from start to finish. This includes developing strategies for:
Explaining trial participation risks in patient-friendly terms
Documenting voluntary consent and re-consent properly
Ensuring comprehension for low-literacy or non-native speakers
Using electronic informed consent (eConsent) platforms compliantly
Addressing site-specific IRB and regulatory variations globally
Every CCRPS certification builds this foundation by embedding informed consent workflows into broader trial responsibilities—whether you're monitoring as a CRA, enrolling patients as a CRC, or leading the trial as a Principal Investigator. This training is fully aligned with GCP guidelines, including ICH E6(R3), and prepares you to manage informed consent in Phase I through Phase IV trials.
You’ll also learn how to avoid common mistakes that trigger audit findings—such as missing signatures, outdated forms, or consent obtained before protocol approval. By emphasizing real-world regulatory readiness, CCRPS ensures that its certified professionals are confident in both paper-based and digital consent systems, across commercial and academic research environments.
Graduates leave the program prepared to answer regulatory queries, pass sponsor audits, and most importantly, ensure participant rights, safety, and understanding. This informed consent training is not an afterthought—it’s an integrated, essential skill embedded throughout CCRPS's clinical research curriculum.
For clinical research professionals looking to advance their careers or meet global compliance standards, mastering informed consent through CCRPS certification is not optional—it’s expected. It’s what separates competent from compliant—and makes you stand out to CROs, sponsors, and regulatory bodies worldwide.
Conclusion
Informed consent in clinical research is not a procedural formality—it is an unwavering commitment to participant autonomy, dignity, and rights. It bridges the gap between ethical research conduct and regulatory compliance, ensuring that participants are fully informed, comprehend the implications of their involvement, and voluntarily decide to participate. This process demands clarity, cultural sensitivity, and continuous engagement, reflecting respect for diverse participant populations. With evolving research protocols, complex study designs, and emerging technologies like eConsent, the responsibility of researchers to uphold robust consent processes grows exponentially.
By mastering the essentials of informed consent, clinical researchers not only safeguard participant welfare but also protect the credibility and integrity of their research. Consent practices must be meticulously planned, thoroughly executed, and continuously updated to align with ethical standards and regulatory frameworks. Ultimately, a strong informed consent process is a declaration of a research team’s commitment to ethical practice. It transforms participants from subjects into empowered partners in advancing science while preserving human dignity and trust.
Frequently Asked Questions
-
Informed consent in clinical research is a legally and ethically required process through which participants voluntarily agree to take part in a study. This agreement follows a comprehensive explanation of the study’s purpose, procedures, risks, benefits, alternatives, data use, and participant rights. Informed consent ensures that participants understand what the study entails and agree without pressure. It’s more than a form; it’s an ongoing dialogue between the researcher and the participant. Key elements include clear communication, comprehension, and voluntariness. Researchers must be proactive in verifying participant understanding, especially in complex or high-risk studies. Consent must be re-obtained if study protocols change or new risk information arises.
-
Informed consent is vital because it upholds participant autonomy and safeguards their rights. Without it, participants might be enrolled in studies without fully understanding the potential risks, benefits, and alternatives. Regulatory bodies such as the FDA, EMA, and IRBs enforce informed consent to ensure ethical conduct and legal compliance. It protects participants from coercion, exploitation, and harm. By obtaining valid consent, researchers demonstrate respect for human dignity and reinforce trust in the research process. Failure to secure proper consent can lead to legal penalties, research invalidation, and loss of public confidence in the study’s integrity.
-
A valid informed consent form in clinical research must include the study’s purpose, procedures, risks, benefits, and available alternatives. It must specify confidentiality measures, data usage, participant rights, and the right to withdraw at any time without penalty. The form should also disclose the researcher’s identity, funding sources, and potential conflicts of interest. Clarity and simplicity are essential, avoiding jargon that could hinder comprehension. Additionally, forms should indicate how new information will be communicated and how to contact the research team for questions. Regulatory bodies require these elements to ensure participants can make informed, voluntary decisions.
-
Ensuring comprehension involves more than providing a consent form. Researchers must engage participants in clear, interactive discussions about the study’s key elements. Techniques such as the teach-back method—where participants explain study details in their own words—are essential. Researchers should provide ample time for questions, assess language proficiency and literacy, and use visual aids if needed. Culturally appropriate communication is crucial. For vulnerable populations, involving family members or legal representatives may enhance understanding. Documentation of these efforts, including participant questions and responses, reinforces the integrity of the consent process and ensures ethical compliance.
-
Yes, certain circumstances allow for waiver or alteration of informed consent, but strict criteria apply. Regulatory bodies like the FDA and IRBs may approve waivers when studies involve minimal risk and when obtaining consent is impractical or could bias the results. Examples include some observational studies or emergency research scenarios. However, such waivers are exceptions, not norms. Researchers must provide justification and obtain approval from regulatory bodies. Even in these cases, participants’ rights and welfare must remain a priority. Transparency, clear communication of the study’s nature, and post-participation debriefing may be required.